Я начну с провокационного заявления — «биологи не публикуют детали своих исследований». Казалось бы столько статей, столько исследований… но где описание и детализация информации, которая получена? Её в принципе нет. А статьи без такой информации пусты и спорны. Каждый нахваливает свой метод, но много ли кто озаботился верификацией чужих данных, а главное смог ли он её сделать?
Можно лишь приветствовать появление таких биоинформационных баз как NCBI genomes и PDB, в которые исследователи помещают данные о секвенированных геномах и структурах РНК, белков. И главное, некоторые ученные прежде чем опубликовать статью, прежде помещают данные в биоинформационные базы.
Вы скажите есть много других баз — но я вам скажу они менее серьезные, и как правило перепосты этих двух с некоторой адаптацией. Но главное, что вся другая биоинформационная информация, можно сказать вторичная — не помещается в базы. А в статьях тем не менее идут различные спекуляции.
Конечно, так оно выглядит только для таких дилетантов как я. У настоящих же профессионалов все как в аптеке. Поэтому можете не утруждать себя ответом на эти пафосные заявления. Мы просто поговорим как выглядит биоинформатика в её частных областях глазами дилетанта. Но может и вас эта история к чему нибудь побудит.
Мы поговорим ниже о построение дерева эволюции согласно Дарвину, посмотрим на сколько это справедливо и таки я в итоге дам полное дерево (в рамках имеющейся информации) эволюции бактерий на основании самых консервативных генов тРНК. И дам пояснение о методе построения такого дерева.
Специалистам в биоинформатике рекомендую читать с раздела №5, пропустив весь мой пафос.
Будучи дилетантом я всегда недоумевал по одному поводу — как можно классифицировать и систематизировать организмы, когда нет информации о ДНК, когда штаммы организмов еще не секвенированы? И пожалуйста Справочник Берджи только в последнем издании начал учитывать информацию о генах. А до этого учитывал только структурные и функциональные особенности бактерий.
Я уже не говорю о тех биологах-консерваторах, которые заявляют на полном серьезе, что таксономия должна строится не только на основании геномного сравнения, но и на основании морфологических и физиологических данных. И это то в генный век мы должны возвращаться во времена К. Линнея?
А ведь при отсутствии более авторитетного издания, чем справочник Берджи, биоинформационные базы по таксономии, такие как в NCBI, хоть и являются более полными и иногда имеют ссылки на секвенированные штаммы — принцип построения такого дерева — это просто перепост справочника Берджи.
Скажите не так… ок, найти отличие можно легко. Но вы никогда не поймете почему дерево именно такое какое оно есть. К тому или иному виду конечно приписано, кто дал такое имя таксону, и если повезет будет статья, и еще если сильно повезет в статье мельком будет описано почему этот таксон поместили так или иначе в систематике.
Дальше если взять отдельные статьи по построению филогенетических деревьев — в них в лучшем случае рассматривается очень небольшое число видов, и строятся деревья совершенно не прозрачными методами и достаточно не большие.
Существует много профессионалов, которые пытаются представить дело так, что проблема дилетанта — это его недообученность и недоосведомленность.
Это отчасти так, но только отчасти. Дилетанты занимаются не своим делом, потому что имея свою профессию — они также интересуются вещами другими и думают, в какой еще сфере они могут применять свои знания. И когда они видят примерно такое состояние как я описал выше для таксономии — они приходят в некоторое замешательство.
Они берут самый наивный метод, так как им нужен результат, а не повод для написания статьи и строят дерево эволюции. Дальше профессионалы начинают возмущаться как же так — они занимаются этим профессионально, а результатов то нет… гранты не все использованы. Хотя можно взять и одному человеку все это построить без особых сложностей и не забивая голову методами, в которых введена сложность ради самой сложности. И вот так получается результат у дилетанта.
Его можно обсуждать, но его можно обсуждать серьезно только тогда, когда у профессионалов будет хоть что-то сравнимое и столь же прозрачное. И вот теперь мы к этому перейдем.
Кто читал мои предшествующие статьи знает, что на эту тему я уже писал начиная со статьи Интересные результаты о эволюционной систематике прокариот или «многовидовое происхождение», и не так давно дал более полные результаты в статье Систематика прокариот — дальние родственники. Здесь я хотел бы рассказать как менялось мое мировоззрение по мере продвижения этого исследования.
Вначале в статье показывалось, что на основании одного вида тРНК, который переносил аланин можно найти устойчивую связь между разными видами, родами и т.д. Эту связь я интерпретировал как половое наследование, т.к. можно было найти организмы у которых существовали тРНК_Ала от двух других видов. Были и некоторые исключения, но их было сравнительно мало. «Что же может быть проще — воскликнул дилетант — это же гены от мамы и папы, и биологи дурят нам голову бесполым размножением».
Эту мысль мои критики почти не заметили тогда (видимо списав на горизонтальный перенос — хотя сильно уж постоянным были связи мама-папа), но отметили что делать выводы на основании одного гена как то не серьезно.
Я охотно согласился, но про себя подумал — а вы то сами сколько генов анализируете? Правильно как правило один 16S, только он подлиннее будет, но зато изрезанный мутациями. Но что нам сравнивать с другими… идем дальше.
Дальше я взял все доступные в NCBI секвенированные геномы и все тРНК и уточнил информацию (см. Систематика прокариот — дальние родственники).
Критики меньше не стало, но она стала больше эмоциональная. Ага, подумал я возражать становится сложнее, а аргументы оппонентов стали далеки от рассматриваемого и косвенные.
Но я видел, что в целом картина стала сильно запутанной, было ощущение, что роды взаимодействуют где-то слабее, где-то сильнее — но почти как каждый с каждым. Тот или иной вид гена у них был общий.
Представить себе, что так могла идти реально эволюция — т.к. как будто все гены бросили в один котел, а потом зачерпывали бы из этого котла случайный набор и создавали вид — было как то сложно. Но результаты говорили об этом неумолимо.
Разные роды хоть и слегка выделялись в группы, но выглядели так как будто тРНК передавались горизонтально случайным образом.
На самом деле 100% оснований не верить в «многовидовое происхождение» нет. Это ровно такая же спекуляция как и дарвиновская эволюция. Эти спекуляции нужно явно называть способами интерпретации данных эксперимента. «Многовидовое происхождение» — это показ графа, на котором есть точные связи по генам между родами.
Но в этом графе нет направления эволюции, этот граф не делает ни каких предположений о прошлом. Он просто показывает факты родства современных организмов. При этом родство этих организмов может быть далеким и на основании этого графа не возможно сказать когда произошла дивергенция видов.
Дарвиновская эволюция — это другой способ интерпретации, который дает возможность наиболее детально представить себе ход эволюции.
Но тут дилетант столкнулся опять с недоумением от классических представлений, а точнее просто от отсутствия результатов. Оппонентом мне было заявлено, что такое понятие как «древний» — плохое для биологии, т.к. на основании имеющихся методов оценить относительное время возникновения видов нельзя. Но мы все таки после уточнения ряда моментов согласились между собой о следующем:
Вот это то я и назвал — интерпретацией по дарвиновской эволюции. Но специально отмечу, что хотя этим то и должны заниматься все дарвинисты (т.е. классические таксономисты и филогенетики), они строят деревья используя меры, которые больше сходны для интерпретации «многовидового происхождения», и конечно им тогда сложно говорить о «древности вида» по определению такой интерпретации — как говорилось выше там нет направления эволюции и не может быть.
Но оппонент оказался не прав в своей оценке «я думаю, что большой разницы между разными видами не будет» — она есть и существенная, это и будет продемонстрировано далее — достаточно посмотреть полученное дерево эволюции.
Отсюда могут читать те, кто брезглив к пафосному тексту дилетанта, который находится выше.
Чтобы понять требуется прочтение статьи Систематика прокариот — дальние родственники, там описаны основы, которые являются входными данными. Поясняя далее, я предполагаю, что вы разобрались, что означает например такой граф и как он был построен:
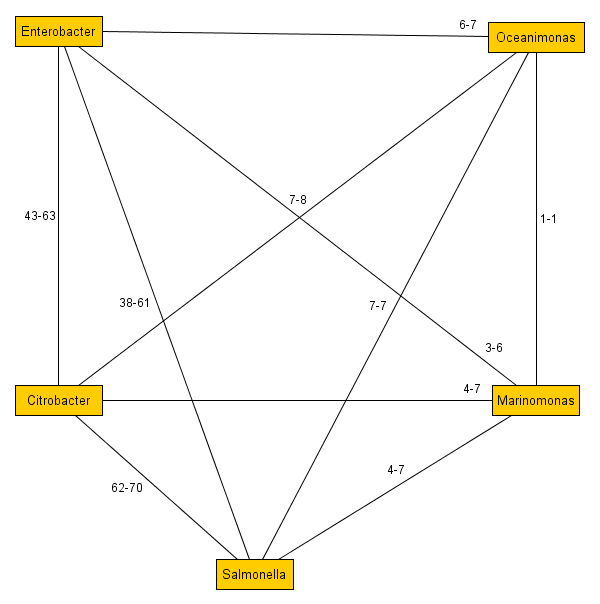
Теперь нам надо разобраться как его преобразовать в дерево с направленной эволюцией, например такое:
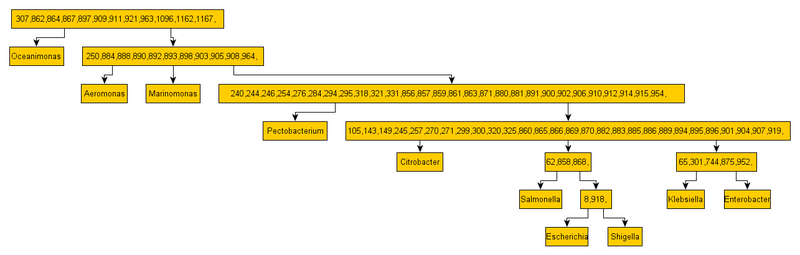
В этом дереве мы восстанавливаем предков современных родов бактерий. Современные рода бактерий имеют названия и представлены как листья дерева, в то время как их предки обозначены набором цифр.
Каждая цифра — это идентификатор группы тРНК, которой обязан обладать предок, чтобы передать своим потомкам в следующие поколение. Если бы он не обладал такой группой тРНК, то мы однозначно не смогли бы получить текущие состояние взаимоотношений (совпадения идентичных тРНК), которое имеется на графе «многовидового происхождения» выше.
Таким образом, алгоритм построения такого дерева состоит из двух частей:
1. Распределение тРНК по группам, так чтобы на всем анализируемом множестве можно было апеллировать только группами без перехода на единичные тРНК — это нужно для двух целей (1) на порядок удобнее иметь дело с группами, чем с большим множеством тРНК. Устраняется дублирующая информация, и группа является минимальной единицей дивергенции. (2) Группы можно отсортировать по количеству тРНК входящих туда. Вероятность дивергенции (разделения) большей группы по разным родам выше при меньшем числе предковых дивергенций (длины ветви).
2. Собственно построение дерева предков.
Далее я опишу только общий принцип реализации эти двух частей.
Разделение на группы:
1. На входе имеется информация вида:
1 10 000913,003420,006818,011215,013800,016316,017374,
2 9 000434,000487,005891,005892,011142,011163,
2 10 000913,003420,006818,007509,011215,013800,016316,017374,
2 8 000487,003420,005891,006678,011163,013218,007509,
она описывает граф «многовидового происхождения», а именно набор связей, где «1» идентификация одного рода, «10» — идентификация второго рода, «000913,003420,006818,011215,013800,016316,017374,» — те тРНК, которые идентичны как в первом, так и во втором роде.
2. Создается первая группа, как набор из всех вообще различных тРНК
3. Происходит распределение по группам, если тРНК на связи между родами относится к группе этот набор заменяется на идентификацию группы, но если вхождение частичное то помечается каких тРНК не хватает, или наоборот какие тРНК, только имеются из этой группы.
4. Разделение группы на две. Анализируется выше сделанное распределение на группы, берется первое частичное вхождение — создается новая группа, а недостающая часть остается у предшествующей группы.
5. Повторяется пункт 3. Так постепенно, произойдет разделение на группы без частичных вхождений.
6. Группы сортируются по величине 1 — группа это набор скажем 20 тРНК, а уже после 300 группы — вхождение 1-2 тРНК
Построение дерева предков:
1. На основании разбиения связей родов по группам тРНК, можно восстановить какие групп тРНК находятся в каждом роде. Так если между родами имеется такая связь
1 10 307|864|867|897|909|911|
6 10 307|862|864|867|897|909|911|
это означает, что группы 307|864|867|897|909|911| есть и у 1-го рода и у 10-го. Но 862 группа к примеру есть только у 10-го и 6-го, но нет у 1-го.
2. Все роды делаем листьями дерева
3. Берем 1-ю группу (помним, что она наиболее крупная, значит она меньше дробилась и является более молодой ). Находим наименьшего общего предка для всех родов, которые обладают этой группой тРНК. Если такого предка нет — создаем его. Если есть, но наименьший общий предок не промаркирован соответствующим идентификатором группы тРНК — маркируем.
4. Повторяем п.3. для всех групп
Ну и собственно результат дерева эволюции бактерий можно посмотреть на картинке:
смотрим в высоком разрешении
P.S. Я понимаю, что я не предоставил собственно результаты разделения на группы, что это за тРНК конкретно понять нельзя, а также метод описан лишь с «птичьего полета». Реально интересующимся я могу предоставить всю информацию, но я жду от них что они попробуют перепроверить меня хоть в чем — то и не постесняются это публично высказать.
Можно лишь приветствовать появление таких биоинформационных баз как NCBI genomes и PDB, в которые исследователи помещают данные о секвенированных геномах и структурах РНК, белков. И главное, некоторые ученные прежде чем опубликовать статью, прежде помещают данные в биоинформационные базы.
Вы скажите есть много других баз — но я вам скажу они менее серьезные, и как правило перепосты этих двух с некоторой адаптацией. Но главное, что вся другая биоинформационная информация, можно сказать вторичная — не помещается в базы. А в статьях тем не менее идут различные спекуляции.
Конечно, так оно выглядит только для таких дилетантов как я. У настоящих же профессионалов все как в аптеке. Поэтому можете не утруждать себя ответом на эти пафосные заявления. Мы просто поговорим как выглядит биоинформатика в её частных областях глазами дилетанта. Но может и вас эта история к чему нибудь побудит.
Мы поговорим ниже о построение дерева эволюции согласно Дарвину, посмотрим на сколько это справедливо и таки я в итоге дам полное дерево (в рамках имеющейся информации) эволюции бактерий на основании самых консервативных генов тРНК. И дам пояснение о методе построения такого дерева.
Специалистам в биоинформатике рекомендую читать с раздела №5, пропустив весь мой пафос.
№1. Таксономия
Будучи дилетантом я всегда недоумевал по одному поводу — как можно классифицировать и систематизировать организмы, когда нет информации о ДНК, когда штаммы организмов еще не секвенированы? И пожалуйста Справочник Берджи только в последнем издании начал учитывать информацию о генах. А до этого учитывал только структурные и функциональные особенности бактерий.
Я уже не говорю о тех биологах-консерваторах, которые заявляют на полном серьезе, что таксономия должна строится не только на основании геномного сравнения, но и на основании морфологических и физиологических данных. И это то в генный век мы должны возвращаться во времена К. Линнея?
А ведь при отсутствии более авторитетного издания, чем справочник Берджи, биоинформационные базы по таксономии, такие как в NCBI, хоть и являются более полными и иногда имеют ссылки на секвенированные штаммы — принцип построения такого дерева — это просто перепост справочника Берджи.
Скажите не так… ок, найти отличие можно легко. Но вы никогда не поймете почему дерево именно такое какое оно есть. К тому или иному виду конечно приписано, кто дал такое имя таксону, и если повезет будет статья, и еще если сильно повезет в статье мельком будет описано почему этот таксон поместили так или иначе в систематике.
Дальше если взять отдельные статьи по построению филогенетических деревьев — в них в лучшем случае рассматривается очень небольшое число видов, и строятся деревья совершенно не прозрачными методами и достаточно не большие.
№2. Проблема дилетанта
Существует много профессионалов, которые пытаются представить дело так, что проблема дилетанта — это его недообученность и недоосведомленность.
Это отчасти так, но только отчасти. Дилетанты занимаются не своим делом, потому что имея свою профессию — они также интересуются вещами другими и думают, в какой еще сфере они могут применять свои знания. И когда они видят примерно такое состояние как я описал выше для таксономии — они приходят в некоторое замешательство.
Они берут самый наивный метод, так как им нужен результат, а не повод для написания статьи и строят дерево эволюции. Дальше профессионалы начинают возмущаться как же так — они занимаются этим профессионально, а результатов то нет… гранты не все использованы. Хотя можно взять и одному человеку все это построить без особых сложностей и не забивая голову методами, в которых введена сложность ради самой сложности. И вот так получается результат у дилетанта.
Его можно обсуждать, но его можно обсуждать серьезно только тогда, когда у профессионалов будет хоть что-то сравнимое и столь же прозрачное. И вот теперь мы к этому перейдем.
№3. Многовидовое происхождение и прочие глупости
Кто читал мои предшествующие статьи знает, что на эту тему я уже писал начиная со статьи Интересные результаты о эволюционной систематике прокариот или «многовидовое происхождение», и не так давно дал более полные результаты в статье Систематика прокариот — дальние родственники. Здесь я хотел бы рассказать как менялось мое мировоззрение по мере продвижения этого исследования.
Вначале в статье показывалось, что на основании одного вида тРНК, который переносил аланин можно найти устойчивую связь между разными видами, родами и т.д. Эту связь я интерпретировал как половое наследование, т.к. можно было найти организмы у которых существовали тРНК_Ала от двух других видов. Были и некоторые исключения, но их было сравнительно мало. «Что же может быть проще — воскликнул дилетант — это же гены от мамы и папы, и биологи дурят нам голову бесполым размножением».
Эту мысль мои критики почти не заметили тогда (видимо списав на горизонтальный перенос — хотя сильно уж постоянным были связи мама-папа), но отметили что делать выводы на основании одного гена как то не серьезно.
Я охотно согласился, но про себя подумал — а вы то сами сколько генов анализируете? Правильно как правило один 16S, только он подлиннее будет, но зато изрезанный мутациями. Но что нам сравнивать с другими… идем дальше.
Дальше я взял все доступные в NCBI секвенированные геномы и все тРНК и уточнил информацию (см. Систематика прокариот — дальние родственники).
Критики меньше не стало, но она стала больше эмоциональная. Ага, подумал я возражать становится сложнее, а аргументы оппонентов стали далеки от рассматриваемого и косвенные.
Но я видел, что в целом картина стала сильно запутанной, было ощущение, что роды взаимодействуют где-то слабее, где-то сильнее — но почти как каждый с каждым. Тот или иной вид гена у них был общий.
Представить себе, что так могла идти реально эволюция — т.к. как будто все гены бросили в один котел, а потом зачерпывали бы из этого котла случайный набор и создавали вид — было как то сложно. Но результаты говорили об этом неумолимо.
Разные роды хоть и слегка выделялись в группы, но выглядели так как будто тРНК передавались горизонтально случайным образом.
№4. Дарвиновская эволюция — как образ мышления
На самом деле 100% оснований не верить в «многовидовое происхождение» нет. Это ровно такая же спекуляция как и дарвиновская эволюция. Эти спекуляции нужно явно называть способами интерпретации данных эксперимента. «Многовидовое происхождение» — это показ графа, на котором есть точные связи по генам между родами.
Но в этом графе нет направления эволюции, этот граф не делает ни каких предположений о прошлом. Он просто показывает факты родства современных организмов. При этом родство этих организмов может быть далеким и на основании этого графа не возможно сказать когда произошла дивергенция видов.
Дарвиновская эволюция — это другой способ интерпретации, который дает возможность наиболее детально представить себе ход эволюции.
Но тут дилетант столкнулся опять с недоумением от классических представлений, а точнее просто от отсутствия результатов. Оппонентом мне было заявлено, что такое понятие как «древний» — плохое для биологии, т.к. на основании имеющихся методов оценить относительное время возникновения видов нельзя. Но мы все таки после уточнения ряда моментов согласились между собой о следующем:
я: О степени консервативности видов можно говорить, как о совокупности наличия более близких к luca консервативных молекул. Вот видимо в чем разница у нас.
оппонент: да, с этим я согласен. то есть если мы можем восстановить «предковое состояние» по большому числу генов (что само по себе задача довольно непростая), то для каждого конкретного вида мы сможем определить, насколько он близок к этому предковому состояния. я думаю, что большой разницы между разными видами не будет, но определенно есть, для кого эволюция шла чуть быстрее, есть те, для кого чуть медленнее. Интуитивно подозреваю, что полученная величина будет очень хорошо коррелировать с длиной каждой конкретной ветви (от корня) для каждого конкретного вида.
Вот это то я и назвал — интерпретацией по дарвиновской эволюции. Но специально отмечу, что хотя этим то и должны заниматься все дарвинисты (т.е. классические таксономисты и филогенетики), они строят деревья используя меры, которые больше сходны для интерпретации «многовидового происхождения», и конечно им тогда сложно говорить о «древности вида» по определению такой интерпретации — как говорилось выше там нет направления эволюции и не может быть.
Но оппонент оказался не прав в своей оценке «я думаю, что большой разницы между разными видами не будет» — она есть и существенная, это и будет продемонстрировано далее — достаточно посмотреть полученное дерево эволюции.
№5. Метод восстановления направления эволюции
Отсюда могут читать те, кто брезглив к пафосному тексту дилетанта, который находится выше.
Чтобы понять требуется прочтение статьи Систематика прокариот — дальние родственники, там описаны основы, которые являются входными данными. Поясняя далее, я предполагаю, что вы разобрались, что означает например такой граф и как он был построен:
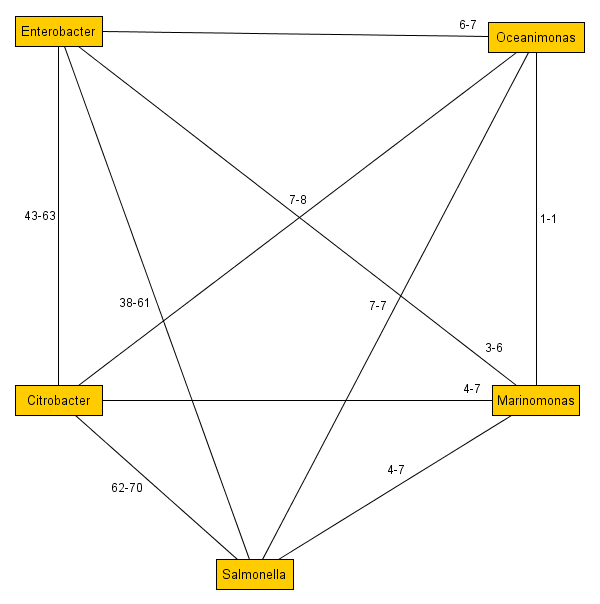
Теперь нам надо разобраться как его преобразовать в дерево с направленной эволюцией, например такое:

В этом дереве мы восстанавливаем предков современных родов бактерий. Современные рода бактерий имеют названия и представлены как листья дерева, в то время как их предки обозначены набором цифр.
Каждая цифра — это идентификатор группы тРНК, которой обязан обладать предок, чтобы передать своим потомкам в следующие поколение. Если бы он не обладал такой группой тРНК, то мы однозначно не смогли бы получить текущие состояние взаимоотношений (совпадения идентичных тРНК), которое имеется на графе «многовидового происхождения» выше.
Таким образом, алгоритм построения такого дерева состоит из двух частей:
1. Распределение тРНК по группам, так чтобы на всем анализируемом множестве можно было апеллировать только группами без перехода на единичные тРНК — это нужно для двух целей (1) на порядок удобнее иметь дело с группами, чем с большим множеством тРНК. Устраняется дублирующая информация, и группа является минимальной единицей дивергенции. (2) Группы можно отсортировать по количеству тРНК входящих туда. Вероятность дивергенции (разделения) большей группы по разным родам выше при меньшем числе предковых дивергенций (длины ветви).
2. Собственно построение дерева предков.
Далее я опишу только общий принцип реализации эти двух частей.
Разделение на группы:
1. На входе имеется информация вида:
1 10 000913,003420,006818,011215,013800,016316,017374,
2 9 000434,000487,005891,005892,011142,011163,
2 10 000913,003420,006818,007509,011215,013800,016316,017374,
2 8 000487,003420,005891,006678,011163,013218,007509,
она описывает граф «многовидового происхождения», а именно набор связей, где «1» идентификация одного рода, «10» — идентификация второго рода, «000913,003420,006818,011215,013800,016316,017374,» — те тРНК, которые идентичны как в первом, так и во втором роде.
2. Создается первая группа, как набор из всех вообще различных тРНК
3. Происходит распределение по группам, если тРНК на связи между родами относится к группе этот набор заменяется на идентификацию группы, но если вхождение частичное то помечается каких тРНК не хватает, или наоборот какие тРНК, только имеются из этой группы.
4. Разделение группы на две. Анализируется выше сделанное распределение на группы, берется первое частичное вхождение — создается новая группа, а недостающая часть остается у предшествующей группы.
5. Повторяется пункт 3. Так постепенно, произойдет разделение на группы без частичных вхождений.
6. Группы сортируются по величине 1 — группа это набор скажем 20 тРНК, а уже после 300 группы — вхождение 1-2 тРНК
Построение дерева предков:
1. На основании разбиения связей родов по группам тРНК, можно восстановить какие групп тРНК находятся в каждом роде. Так если между родами имеется такая связь
1 10 307|864|867|897|909|911|
6 10 307|862|864|867|897|909|911|
это означает, что группы 307|864|867|897|909|911| есть и у 1-го рода и у 10-го. Но 862 группа к примеру есть только у 10-го и 6-го, но нет у 1-го.
2. Все роды делаем листьями дерева
3. Берем 1-ю группу (помним, что она наиболее крупная, значит она меньше дробилась и является более молодой ). Находим наименьшего общего предка для всех родов, которые обладают этой группой тРНК. Если такого предка нет — создаем его. Если есть, но наименьший общий предок не промаркирован соответствующим идентификатором группы тРНК — маркируем.
4. Повторяем п.3. для всех групп
Ну и собственно результат дерева эволюции бактерий можно посмотреть на картинке:
смотрим в высоком разрешении
P.S. Я понимаю, что я не предоставил собственно результаты разделения на группы, что это за тРНК конкретно понять нельзя, а также метод описан лишь с «птичьего полета». Реально интересующимся я могу предоставить всю информацию, но я жду от них что они попробуют перепроверить меня хоть в чем — то и не постесняются это публично высказать.
Only registered users can participate in poll. Log in, please.
Состав статьи
51.65% Статья в целом интересна94
15.38% Если не читать первые пафосные 4 раздела, то интересно28
32.97% Пафосные 4 раздела имеют важное значение для статьи60
182 users voted. 132 users abstained.

{kind=link}