 Статья 2013-ого года «A support vector machine for identification of single-nucleotide polymorphisms from next-generation sequencing data» (O'Fallon, Wooderchak-Donahue, Crockett) предлагает новый метод определения полиформизмов в геноме на основе применения метода опорных векторов (SVM). Хотя ранее в статье 2011-ого года «A framework for variation discovery and genotyping using next-generation DNA sequencing data» уже описывалось применение методов машинного обучения для определения однонуклеотидных полиморфизмов (SNP-ов, снипов), подход, основанный на использовании SVM, описан впервые в данной статье.
Статья 2013-ого года «A support vector machine for identification of single-nucleotide polymorphisms from next-generation sequencing data» (O'Fallon, Wooderchak-Donahue, Crockett) предлагает новый метод определения полиформизмов в геноме на основе применения метода опорных векторов (SVM). Хотя ранее в статье 2011-ого года «A framework for variation discovery and genotyping using next-generation DNA sequencing data» уже описывалось применение методов машинного обучения для определения однонуклеотидных полиморфизмов (SNP-ов, снипов), подход, основанный на использовании SVM, описан впервые в данной статье.Определение полиморфизмов в геноме является важной (например, для полногеномного поиска ассоциаций aka GWAS), но нетривиальной задачей. Приходится учитывать, что многие организмы гетерозиготны, а также, что данные могут содержать ошибочную информацию.
Стандартный подход к определению SNP основан на выравнивании данных секвенирования (ридов, фрагментов генома) относительно референсного (эталонного) генома. Однако риды могут содержать ошибки и могут быть неправильно выровнены на референс. Поскольку считается, что возникновение ошибки в данной позиции – событие более редкое, чем появление полиморфизма, наиболее простым подходом в данном случае было бы отфильтровать варианты с маленькой частотой. Однако в статье 2010-ого года «Uncovering the roles of rare variants in common disease through whole-genome sequencing» было показано, что наибольший эффект на многие заболевания имеют редкие полиморфизмы, возникающие с частотой < 1%. Фильтрация на основе частоты повлечёт за собой большое количество false negatives для редких SNP, поэтому возникает необходимость в создании более чувствительных методов определения геномных вариаций.
Как указано в статье, преимущество методов, основанных на машинном обучении, состоит в том, что они позволяют комбинировать разные факторы, влияющие на правдоподобность возникновения полиморфизма в данной позиции в геноме, что в том числе повышает чувствительность метода к более редким полиморфизмам.
В отличие от других методов машинного обучения (например, от подхода, основанного на обучении с подкреплением, описанного в вышеуказанной статье), SVM требуется значительно меньшее количество данных для обучения.
В рассматриваемой статье не описывается подробный метод реализации алгоритма, но даются ссылки на другие статьи, содержащие технические детали. В качестве платформы, предоставляющией реализацию алгоритма SVM, используется LIBSVM. Первая публикация о LIBSVM относится к 2011 году, а последний релиз на сайте — к 2013. Необходимо отметить, что в качестве входных данных принимаются файлы в формате SAM или BAM, что кажется удобным и естественным подходом в биоинформатике. В результате работы алгоритма генерируется файл формата VCF.
Для обучения модели используются данные двух типов:
- выравнивания, соответствующие истинным полиморфизмам;
- несовпадения нуклеотидов в определенных позициях, не связанных с наличием полиморфизмов.
Интересно, как можно собрать данные, соответствующие второму типу. Авторы приводят в пример две возможные стратегии:
- Определить полиморфизмы с помощью GATK’s best practices guidelines, включая UnifiedGenotyper. Авторы использовали снипы, которые определились только в одном случае (при этом исключив снипы, которые описаны в данных проекта 1000 Genomes), из оставшихся выбрали снипы с очень низким значением качества выравнивания.
- Авторы выбрали полиморфизмы, которые встречаются хотя бы 8 раз в локальной базе 57 экзомов (которые также не соответствуют данным из 1000 Genomes).
Следует отметить, что полиморфизмы могут встречаться также в нетранскрибируемых участках генома, поэтому, возможно, использование информации, полученной исключительно из анализа экзомов, может повлечь некоторые неточности.
На полученных данных производится обучение. В статье указано, что обучение включает определение численных параметров гиперплоскости, наилучшим образом разделяющей два класса данных, на которых производится обучение.
Интересно также, какие признаки были выбраны авторами для обучения модели. Авторы обучали модель, начиная с множества, состоящиго из трех признаков. Это давало плохой результат, поэтому множество признаков расширяли. Этот процесс описан в статье не очень подробно, и не совсем понятно, в каком порядке авторы добавляли новые признаки, ведь очевидно, что они не перебирали все существующие подмножества признаков. Итого, авторами были рассмотрены 16 признаков, среди них вероятность ошибки в ридах, среднее качество данного нуклеотида в ридах, сумма качеств нуклеотидов в данной позиции (последние два не дали никакого улучшения при добавлении в множество признаков), качество выравнивания ридов, глубина секвенирования (последнее также не дало улучшения работы модели), баланс аллелей, качество рядом расположенных нуклеотидов.
Согласно графику, приведенному в статье, описанный метод превосходит другие методы определения полиморфизмов по чувствительности и специфичности на определенном датасете NA12878. Метод определил 95.6% всех true positives, в то время как другие методы: UnifiedGenotyper, UnifiedGenotyper+VQSR, SAMtools и FreeBayes определили лишь 90.6%, 94.1%, 89.5% и 88.7% соответственно.
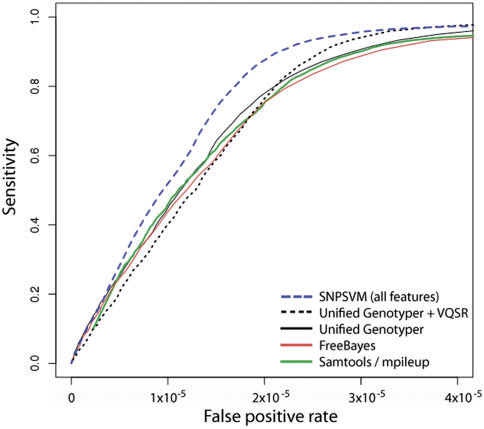
Среди недостатков описанного подхода можно заметить, что модель SVM может давать плохие результаты на данных, содержащих разные профили ошибок (error profile). Кроме того, несмотря на многопоточность, реализация оказалась на 10-20% медленнее, чем метод GATK UnifiedGenotyper.
В целом выбранная модель достаточно хорошо соответствует специфике задачи и соответствует общей тенденции использования методов машинного обучения для анализа геномных последовательностей.
Статья написана в рамках семинара по биоинформатике.
