Комментарии 75
Тема интересная, правда я мало чего понял :)
После прочтения возник дилетанский вопрос — вот вы рассматриваете начальные состояния и способы сворачивания из них. И получается, что все это влияет на конечный результат. А можно ли взять задачу наоборот — взять конечное состояние (как я понимаю, это уже готовый свернутый белок), оно уже известно, т.к. структуры белков изучены и смоделировать процесс «разворачивания», к примеру поменяв знак притягивания-отталкивания. Таким образом получим начальное состояние и посмотреть что оно из себя представляет, вытянутая линия ли это или клубок.
После прочтения возник дилетанский вопрос — вот вы рассматриваете начальные состояния и способы сворачивания из них. И получается, что все это влияет на конечный результат. А можно ли взять задачу наоборот — взять конечное состояние (как я понимаю, это уже готовый свернутый белок), оно уже известно, т.к. структуры белков изучены и смоделировать процесс «разворачивания», к примеру поменяв знак притягивания-отталкивания. Таким образом получим начальное состояние и посмотреть что оно из себя представляет, вытянутая линия ли это или клубок.
Здесь мы говорим о РНК, а не белках. Почему — Часть №1. Введение в биовычисления по сворачиванию. От белков к РНК. Но проблемы конечно сходные, просто у белков будет сложнее.
Надо понимать, что вытянутая линия в природе не бывает — это идеализация. Цель этой идеализации было посмотреть, а есть ли вообще разница в начальном положении. Получается, что есть, хотя может оказаться, что можно свернуть достаточно далеко — и лишь потом, неправильное начальное положение окажет своей действие.
С другой стороны, если сворачивать вообще из начального положения клубок, но случайного — то будет еще сложнее — цепь просто не развернется, чтобы свернуться.
Средний вариант я как раз тут и описываю, и почему я к нему пришел.
Что касается вашего вопроса, а если сделать наоборот? С одной стороны, конечно это можно сделать — причем развернуть легче. Но также существует множество вариаций это сделать. И в отличии от процесса «от начального к конечному» — мы не будем знать точно критерии к чему стремится. Т.е. будет еще сложнее.
Надо понимать, что вытянутая линия в природе не бывает — это идеализация. Цель этой идеализации было посмотреть, а есть ли вообще разница в начальном положении. Получается, что есть, хотя может оказаться, что можно свернуть достаточно далеко — и лишь потом, неправильное начальное положение окажет своей действие.
С другой стороны, если сворачивать вообще из начального положения клубок, но случайного — то будет еще сложнее — цепь просто не развернется, чтобы свернуться.
Средний вариант я как раз тут и описываю, и почему я к нему пришел.
Что касается вашего вопроса, а если сделать наоборот? С одной стороны, конечно это можно сделать — причем развернуть легче. Но также существует множество вариаций это сделать. И в отличии от процесса «от начального к конечному» — мы не будем знать точно критерии к чему стремится. Т.е. будет еще сложнее.
Впрочем, я еще раз подумал. Мы можем определить геометрическую энтропию как желание увеличить свою свободу произвольно вращаться. Тогда если уже в свернутой цепи, начать постепенно увеличивать такую энтропию — то мы сможем выявить те нуклеотиды «слабые звенья», которые потеряют водородные связи вначале, а что затем. Это произойдет не сразу, и все же будут различия (не все связи порвутся одновременно). Затем получится некое промежуточное состояния, в котором, скорее всего, еще будут видны зачатки воздействия стэкинга, но с увеличением энтропии и это потеряется.
Это надо проверять, но похоже это будет работать лишь в другом направлении как сворачивание, принципы которого я изложил в этой статье — в выводах.
Это надо проверять, но похоже это будет работать лишь в другом направлении как сворачивание, принципы которого я изложил в этой статье — в выводах.
НЛО прилетело и опубликовало эту надпись здесь
???
Видимо имелась цель сказать, что чувак ничего не понял из статьи, и я его понимаю. я тоже после прочтения почувствовал себя дикарем.
Наверное полезно тогда начать с Часть №1. Введение в биовычисления по сворачиванию. От белков к РНК. К сожалению, есть некоторый порог вхождения в тему — как и везде впрочем, но на мой взгляд, — оно того стоит. Я тоже года 4 назад вообще ничего не понимал в этом, и даже книги в этом мало помогали.
Ну, или может есть конкретные вопросы? Что именно не ясно?
Рекомендую просто при написании серийных постов после долгого перерыва приводить в шапке ссылки на предыдущие статьи цикла. Тогда читатель сразу увидит, что он не «дикарь», а просто тема специализированная.
В прошлых статьях, я это делал. Но тут я хотел как раз подчеркнуть, что тем не столь специализированна как кажется. И для понимания этой статьи не обательно прочтение чего-то еще. Минимум нужны знания, описанные в первой части — по сути знание школьной биологии.
По сути же тут нет ничего специального — смотрим на картинки и все понимаем :) Не, ну честно… единственно возможно есть несколько непривычных терминов, но необходимы я же поясняю…
Лично мне все понятно и интересно — это просто было пожелание для удобства ориентирования не привыкших еще юзеров. Сам пока еще не спец, но присматриваюсь к этой теме на дальнюю перспективу с позиции широкопрофильного ИТшника: что можно будет сделать полезного в ближайшие годы в плане железа и софта, чтобы это вызвало какую-нибудь disruptive инновацию в биологии; что для этого актуальнее всего учить, каковы перспективы в этой сфере у стартапов или краудфандинговых инициатив и т. д.
НЛО прилетело и опубликовало эту надпись здесь
Я честно говоря, не вправе отвечать на такие вопросы, я не биолог. Но вот, что мне кажется, такие белки — это скорее крупный функциональный механизм. Эти белки имеют все равно участки, которые стабильно сворачиваются. И совсем другое дело, что под действием некоторых стимулов (контактов с другими белками/РНК, атомами и т.д.) они начинаю работать, изменяя свою конформацию. У этих белков видимо степень изменения достаточно большая.
К вашим вопросам:
1. У РНК все несколько проще, скорее из-за их не большой длины. Но вот чем интересен именно этот рибозим, сворачивание которого я тут описывал. Он имеет замечательное свойство саморазрезаться. После чего эти части аналогично комплементарным связям как в ДНК достараиваются — и в итоге происходит размножение. Как я понимаю вопрос о сшивке остается открытым, но тем не менее достоверно известно, что такие РНК саморазмножаются. Таким образом, можно говорить о наименьшей «живой» молекуле.
Так вот такие свойства это РНК конечно не могут получится в статике. Не закрепленные никакими связями отдельные нуклеотиды продолжают флуктуировать. Но в данном рибозиме, все связи образованны так, что они специально подталкивают крайнею близость «выступа соединяющего спирали L1 и L2 (по центру сверху)» с одним из концов спирали — это место как раз место разреза.
2. Поэтому то, что мне не удается смоделировать сворачивание концов — можно еще объяснить тем, что в этот момент на самом деле происходит разрезание. А биологи получают состояние уже после разрезания. Т.е. само сворачивание приводит к тому, что молекула разрезается.
К сожалению точных критериев тут нет. Сейчас кажется, что это ошибка — и вообще-то надо проверить другие варианты — все возможное поле вариаций. Только тогда можно понять, что в реальности происходит что-то такое, что не учитывается в моделировании. Причем надо показать, что это происходит с необходимостью.
3. Какие способы? Как можно понять из моих статей, одна из претензий к существующему подходу — это как раз использование случайности и статистического подхода. Я против такого подхода, т.к. уверен, что он совершенно не характеризует состояние макромолекулы. Силы и состояния в молекулы, совершенно разные не только в разные моменты, но и в каждом месте. Поэтому моделировать это можно лишь учитывая различия происходящие в разных местах.
Нужно определять не обще статистические параметры молекулы, как например, достижение минимальной энергии. А такие параметры, которые характеризуют сворачивание в отдельных местах — наличие стэкинга, стремление к образованию водородных связей и т.д. Когда это выполняется, то можно быть уверенным, что сворачивание произошло верно. Это не будет выполняться — если чего-то не хватает для сворачивания. К примеру, для образования отдельных спиралей — совершенно не важно учитывать стэкинг, но для многоспиральных это становится важным. Так вот для таких «динамичных» белков ( IDPs) или близких аналогов РНК — может быть важно учитывать что-то еще. Как правило тогда появляются как раз те стимулы-активаторы (медиаторы, их кажется называют), которые и управляют «динамикой» сворачивания/разворачивания. И вот их и нужно думать как учитывать в моделировании… но это будет еще сложнее, чем то что я пытаюсь сейчас сделать.
К вашим вопросам:
1. У РНК все несколько проще, скорее из-за их не большой длины. Но вот чем интересен именно этот рибозим, сворачивание которого я тут описывал. Он имеет замечательное свойство саморазрезаться. После чего эти части аналогично комплементарным связям как в ДНК достараиваются — и в итоге происходит размножение. Как я понимаю вопрос о сшивке остается открытым, но тем не менее достоверно известно, что такие РНК саморазмножаются. Таким образом, можно говорить о наименьшей «живой» молекуле.
Так вот такие свойства это РНК конечно не могут получится в статике. Не закрепленные никакими связями отдельные нуклеотиды продолжают флуктуировать. Но в данном рибозиме, все связи образованны так, что они специально подталкивают крайнею близость «выступа соединяющего спирали L1 и L2 (по центру сверху)» с одним из концов спирали — это место как раз место разреза.
2. Поэтому то, что мне не удается смоделировать сворачивание концов — можно еще объяснить тем, что в этот момент на самом деле происходит разрезание. А биологи получают состояние уже после разрезания. Т.е. само сворачивание приводит к тому, что молекула разрезается.
К сожалению точных критериев тут нет. Сейчас кажется, что это ошибка — и вообще-то надо проверить другие варианты — все возможное поле вариаций. Только тогда можно понять, что в реальности происходит что-то такое, что не учитывается в моделировании. Причем надо показать, что это происходит с необходимостью.
3. Какие способы? Как можно понять из моих статей, одна из претензий к существующему подходу — это как раз использование случайности и статистического подхода. Я против такого подхода, т.к. уверен, что он совершенно не характеризует состояние макромолекулы. Силы и состояния в молекулы, совершенно разные не только в разные моменты, но и в каждом месте. Поэтому моделировать это можно лишь учитывая различия происходящие в разных местах.
Нужно определять не обще статистические параметры молекулы, как например, достижение минимальной энергии. А такие параметры, которые характеризуют сворачивание в отдельных местах — наличие стэкинга, стремление к образованию водородных связей и т.д. Когда это выполняется, то можно быть уверенным, что сворачивание произошло верно. Это не будет выполняться — если чего-то не хватает для сворачивания. К примеру, для образования отдельных спиралей — совершенно не важно учитывать стэкинг, но для многоспиральных это становится важным. Так вот для таких «динамичных» белков ( IDPs) или близких аналогов РНК — может быть важно учитывать что-то еще. Как правило тогда появляются как раз те стимулы-активаторы (медиаторы, их кажется называют), которые и управляют «динамикой» сворачивания/разворачивания. И вот их и нужно думать как учитывать в моделировании… но это будет еще сложнее, чем то что я пытаюсь сейчас сделать.
А GROMACS вы в своих работах не думали использовать?
А чем он мне может помочь? Имеется ли у GROMACS открытый код? Если нет как его можно использовать, если не совершенствовать?
Ну и потом там используется т.н. «молекулярная динамика», что вообще-то я своим подходом — критикую, как излишнее усложненный и не практичный подход к моделированию :) (впрочем, если бы кто-то на пальцах это могу бы доступно рассказать и пояснить, чем «молекулярная динамика» хороша — можно было бы подискутировать, но копать в этом направлении самому — считаю малоинтересным и не перспективным)
И если, что-то использовать от молекулярной динамики — то NAMD, т.к. визуализатор VMD — я как раз уже переписал и использую как отображение графики (см. История одного реинжиниринга или RNAInSpace v.1.3. Demo)
Ну я просто искал что-нибудь, чтобы моделировать мицеллообразование небелковых молекул (включая кулоновские взаимодействия ионов по particle mesh evald), и пока мне импонирует именно gromacs. Да, это GPL-программа, есть в репозиториях любого дистрибутива, один из самых быстрых пакетов молекулярного моделирования, есть встроенный визуализатор и пакеты для анализа полученных данных, списки рассылки проекта, и gromacs manual с описанием всех алгоритмов и т.д. и т.п. Смоделировать мне нужно, к примеру, взаимодействие этого: 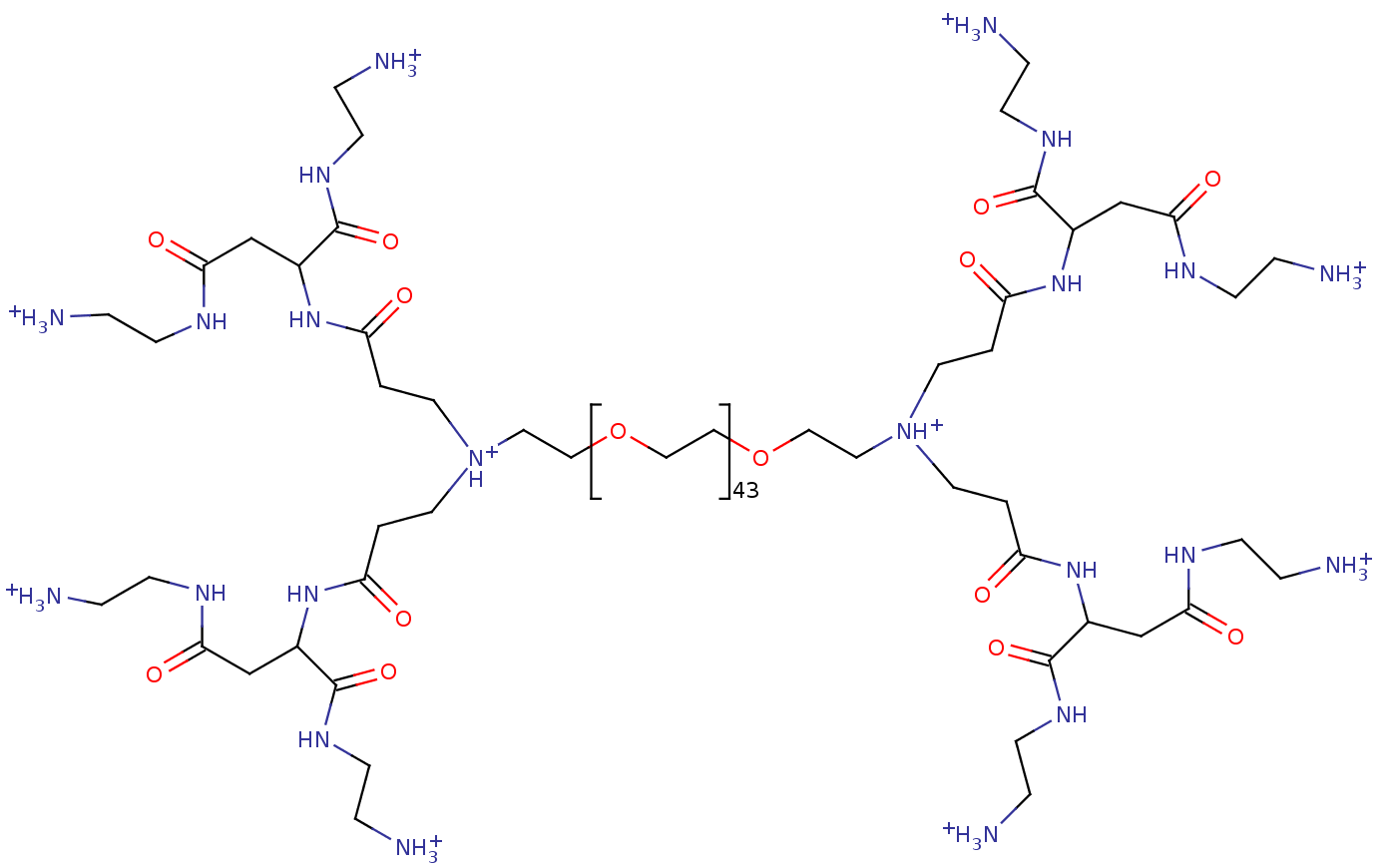
с этим:
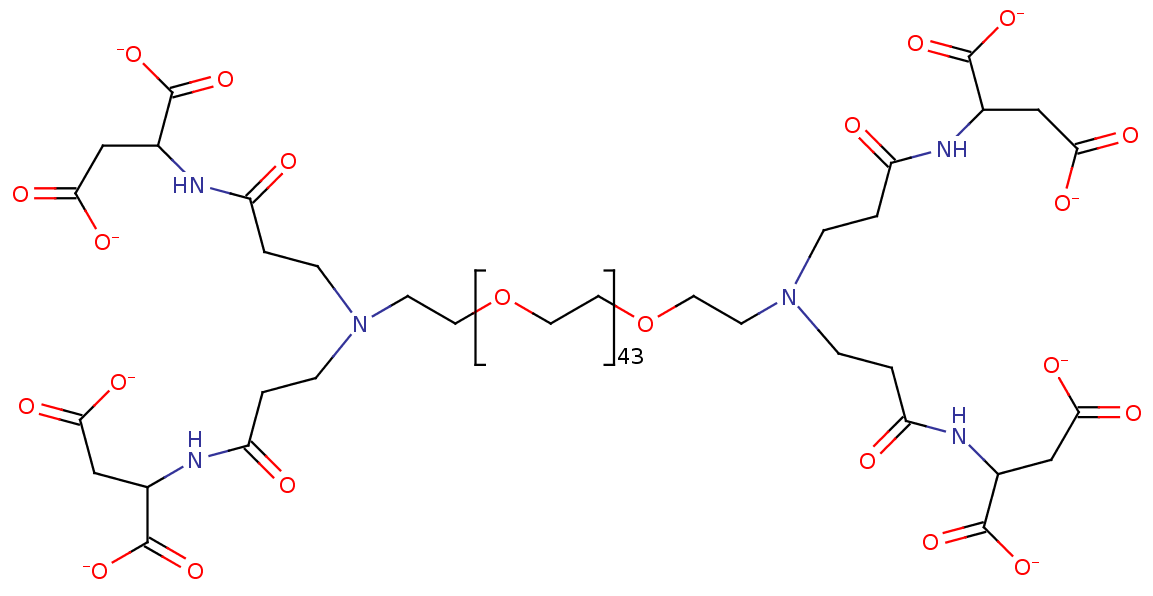
При этом важно учесть влияние растворителей, потому что в хлороформе они дают устойчивый трехмерный гель, а в воде и метаноле этого не наблюдается, что логично, учитывая сольватацию ионов и разделение ионных пар. Будет ли лучше использовать NAMD для этого?

с этим:

При этом важно учесть влияние растворителей, потому что в хлороформе они дают устойчивый трехмерный гель, а в воде и метаноле этого не наблюдается, что логично, учитывая сольватацию ионов и разделение ионных пар. Будет ли лучше использовать NAMD для этого?
В этом я ориентируюсь как раз плохо. Получилось смоделировать? может напишите на хабре статью? Было бы интересно…
Пока что я только получил заряды на всех атомах с помощью французского R.E.D.-сервера, теперь буду пытаться перевести мои молекулы в понятный gromacs вид (они ведь небелковые, будут грабли) и начинать думать над параметрами моделирования. Gromacs вроде более-менее законченный пакет, меня как непрограммиста он пугает менее всего. Если все получится, то обязательно напишу статью.
Уважаемый tac, не поймите как поклеп лично на вас, НО, по фолдингу РНК ТЬМА литературы, просто огромное количество! теперь вы говорите о учете стэкинга, потом вы начнете говорить о учете других параметров, в итоге придете к комбинации силового поля и статистических потенциалах, о которых я вам говорил 3 поста назад.
Продолжаю — вам будет необходимо силовое поле (force field), которые будет себя вести адекватно данной биологической мишени, универсального пока не создали. Оно должно учитывать все вероятные взаимодействия между RNA — водородные связи, ван-дер-Ваальсовы взаимодействия, pi-pi и катион/анион-pi стэкинги, а если там еще и метал есть, который стабилизирует фолдинг, то добавьте еще специфических взаимодействий.
Литература
К примеру смотрите сюда
DNA Polymorphism: A Comparison of Force Fields for Nucleic Acids
www.cell.com/biophysj/abstract/S0006-3495(03)74957-1
или сюда
RNA Folding: Conformational Statistics, Folding Kinetics, and Ion Electrostatics
www.annualreviews.org/doi/abs/10.1146/annurev.biophys.37.032807.125957?journalCode=biophys
Продолжаю — вам будет необходимо силовое поле (force field), которые будет себя вести адекватно данной биологической мишени, универсального пока не создали. Оно должно учитывать все вероятные взаимодействия между RNA — водородные связи, ван-дер-Ваальсовы взаимодействия, pi-pi и катион/анион-pi стэкинги, а если там еще и метал есть, который стабилизирует фолдинг, то добавьте еще специфических взаимодействий.
Литература
К примеру смотрите сюда
DNA Polymorphism: A Comparison of Force Fields for Nucleic Acids
www.cell.com/biophysj/abstract/S0006-3495(03)74957-1
или сюда
RNA Folding: Conformational Statistics, Folding Kinetics, and Ion Electrostatics
www.annualreviews.org/doi/abs/10.1146/annurev.biophys.37.032807.125957?journalCode=biophys
В том то и дело, мне не понадобится НИКАКА физика или химия. О водородных связях и стэкинге речь идет только в терминах геометрического расположения молекул.
Литература говорите, вы мне лучше покажите программу которая сворачивает РНК, причем только с вводом первичной последовательности и предполагаемых водородных связей. Зачем мне читать сказки на ночь, которые не приближают к результату не на грамм? Для общего развития, спасибо — лучше фантастику Лема почитаю.
не моя специальность, но мне кажется пока это маловероятно :) копипастните сюда последовательность (без водородны связей) — попробую на доступных программах
Спасибо.
aagaggucggcaccugacgucgguguccugaugaagauccaugacaggaucgaaaccucuu
Расскажите, даже если будет отрицательный результат. А лучше статью на хабре :)
aagaggucggcaccugacgucgguguccugaugaagauccaugacaggaucgaaaccucuu
Расскажите, даже если будет отрицательный результат. А лучше статью на хабре :)
статья к сожалению отнимает тьму времени :( минимум час
Ну, это же не сравнимо если пробовать получить результат на «доступных программах», кстати, что вы собираетесь использовать?
использовать — все доступное
мне необходимы условия
1. Последовательность (уже есть)
2. С какой структурой сравнивать — тк критерий сворачивания это RMSD — среднеквадратичное отклонение.
мне необходимы условия
1. Последовательность (уже есть)
2. С какой структурой сравнивать — тк критерий сворачивания это RMSD — среднеквадратичное отклонение.
Тут у вас будут проблемы, впрочем…
Давайте возьмем, ту с которой сравниваю я интеллектуально (моя программа об этом не знает) — 2QUS
Давайте возьмем, ту с которой сравниваю я интеллектуально (моя программа об этом не знает) — 2QUS
2QUS — это то, что вы пытаетесь свернуть?
Нет, это то с чем сравнивать! То, что я пытаюсь свернуть в PDB нет.
а как вы будете оценить «правильность» сворачивания? одна-две базу могут «повернуть» сворачивание совсем по другому.
Вы вначале дайте хоть какой-то вариант — я оценю визуально. И это будет супер защита от мухлежа подобных алгоритмов.
Знаете это похоже вот на что, из области АИ:
1. предлагается конструкция машины, предназначенной для моделирования человеческого мозга, который не описан;
2. подробно описанные характеристики машины полагаются аналогичными характеристикам мозга;
3. затем делается «открытие», что машина ведет себя подобно мозгу;
порочность состоит в «открытии» того, что было постулировано".
Знаете это похоже вот на что, из области АИ:
1. предлагается конструкция машины, предназначенной для моделирования человеческого мозга, который не описан;
2. подробно описанные характеристики машины полагаются аналогичными характеристикам мозга;
3. затем делается «открытие», что машина ведет себя подобно мозгу;
порочность состоит в «открытии» того, что было постулировано".
Зачем Вам по ходу решения знать готовое решение? Я чувствовал не доброе, но похоже оно реально так пахнет…
Я понял. Вы наверное это не сможете :)
Хорошо, давайте тогда смоделируем 2QUS — попробуйте её получить in silico, правда там не приятность в PDB файле нет водородов, поэтому лучше использовать первичную последовательность, по которой достроить водороды.
Хорошо, давайте тогда смоделируем 2QUS — попробуйте её получить in silico, правда там не приятность в PDB файле нет водородов, поэтому лучше использовать первичную последовательность, по которой достроить водороды.
нет, уж :) давайте сами!
в wiki куча ссылок, я думаю вам будет интересно проверить сначала литературные данные и известные алгоритмы, чем городить лес абстракций. А то это получается псевдонаука, а не наука
в wiki куча ссылок, я думаю вам будет интересно проверить сначала литературные данные и известные алгоритмы, чем городить лес абстракций. А то это получается псевдонаука, а не наука
Нет. Это интересно мало. Наука не заключается в том, чтобы проверять все абсурдные варианты, которые не дают результатов.
Я пас. То, что вы делаете к науке имеет слабое отношение.
Это увы, ваше сугубо частное мнение.
Да я понял, что вы пас, т.к. не получится вам продемонстрировать результат того, что я вас прошу. Но с пеной у рта, будем доказывать, что есть куча литературы, замечательных алгоритмов и т.д. — Результаты где? Это вот как раз и не наука — а лишь публикация сказок.
Хотя странно, что как критерий используется среднеквадратичное отклонение от того, что знаем. Такое великое поле для мухлежа- идем и стремимся достичь те углы, которые есть у образца — что может быть проще :)
Ну и к сведению, что
что хорошо согласуется с моими выводами, которые я раньше тут излагал.
Поэтому не стоит тут вести разговор ad hominem, и решать что есть наука, а что нет. Может надо просто разобраться в теме и угле зрения о котором тут говорится, и обновить свои устаревающие знания?
Считается, что величина RMSD не может служить достоверным показателем степени свернутости биологической молекулы. В данной работе авторы применительно к РНК предложили использовать величину «нативная характеристика» — NC, которая была определена как разница количества межатомных контактов, присутствующих в нативной конформации и количество контактов, не характерных для нативной конформации
Does Water Play a Structural Role in the Folding of Small Nucleic Acids?
что хорошо согласуется с моими выводами, которые я раньше тут излагал.
Поэтому не стоит тут вести разговор ad hominem, и решать что есть наука, а что нет. Может надо просто разобраться в теме и угле зрения о котором тут говорится, и обновить свои устаревающие знания?
НЛО прилетело и опубликовало эту надпись здесь
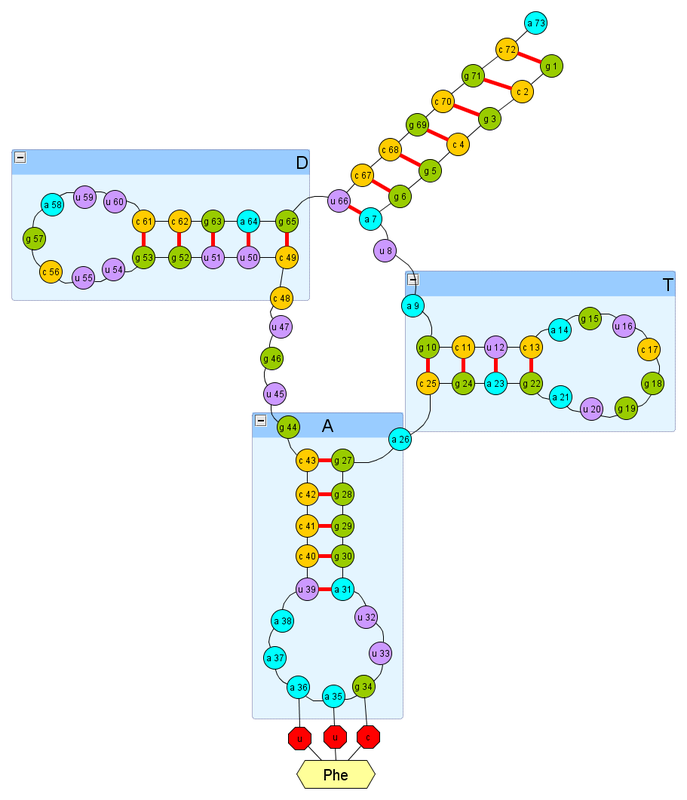
Вообще-то да, прошу дать трехмерную модель. Что касается двумерной — у вас она получилась не верна —
правильная
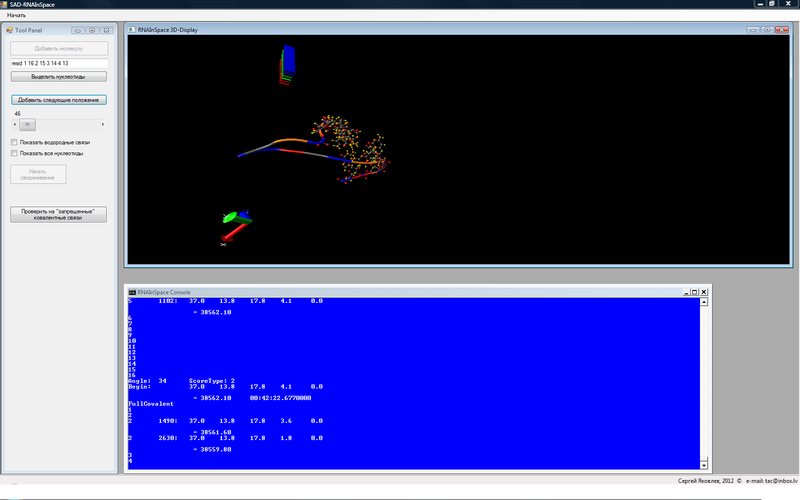
кроме того на ней не отображены еще неканонические пары —
a44 — u15
a44 — u20
с28-g33
не знаю что вы использовали, но двумерные модели хорошо дает RNAfold web server
но он конечно дает только каннонические пары.
правильная
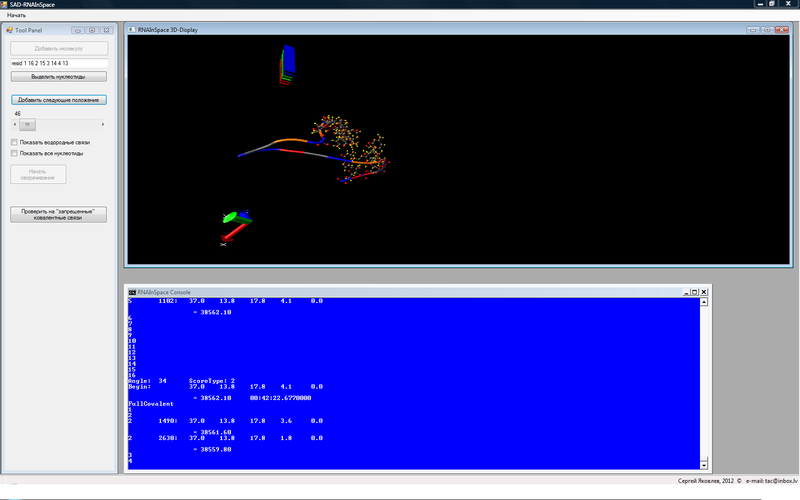
кроме того на ней не отображены еще неканонические пары —
a44 — u15
a44 — u20
с28-g33
не знаю что вы использовали, но двумерные модели хорошо дает RNAfold web server
но он конечно дает только каннонические пары.
Блин, ошибся ссылкой
Причем ошибся ДВАЖДЫ, извиняюсь
вот правильная вторичная структура

вот правильная вторичная структура
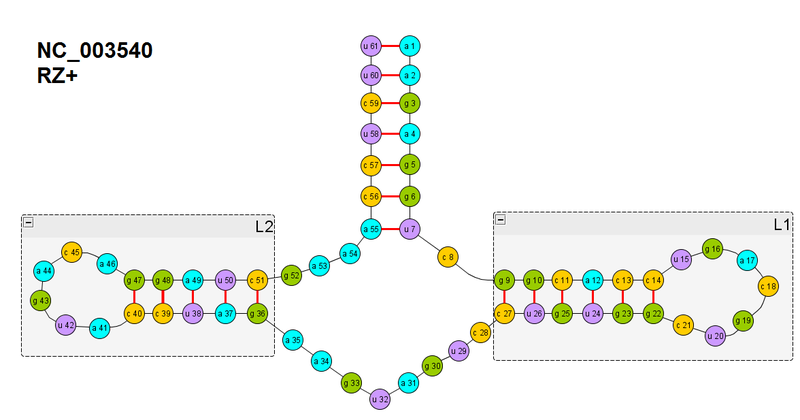
> Думаю проработать вашу идею и через неделю написать что-то по существу, если получится.
Спасибо за интерес, как на счет поучаствовать в этом несколько глубже? На раз тут сложно, что-то сделать… не хотите попробовать демо моей программы?
Спасибо за интерес, как на счет поучаствовать в этом несколько глубже? На раз тут сложно, что-то сделать… не хотите попробовать демо моей программы?
Но похоже вы опять читали, по горизонтали. Да, я говорю о учете стэкинга. Но и более того, я говорю какую роль играет стэкинг в процессе сворачивания. Покажите мне хоть одну книгу/статью где об этом говорится?
Base-stacking and base-pairing contributions into thermal stability of the DNA double helix
nar.oxfordjournals.org/content/34/2/564.short
это первое что под руку подвернулось
nar.oxfordjournals.org/content/34/2/564.short
это первое что под руку подвернулось
И потом, самое главное осознать, что задача фолдинга — это не задача физики или химии :) Что и отличает мой подход — нет ничего кроме углов и расстояний, и правил какими они должны быть и стремится. По сути это задача конструирования механизмов, причем конструирования именно в плане соединения деталей. И задача как бы стоит достаточно обще — как вообще можно проектировать вариации соединений, чтобы их не разрушила энтропия. Конечно, это фантастика сейчас… но реальность делаем мы :)
Причем энтропия в этом случае — это совсем не физическая сущность, а геометрическая. Мера того на сколько можно провернуться в пространстве не столкнувшись с другими объектами.
Т. е. в РНК нет своих аналогов гидрофилии и гидрофобии, как в Foldit, тут все решают только внутренние связи, которые зависят от кода? т. е. «петли» внешними сторонами друг на друга никак не влияют, кроме того, что физически занимают пространство?
Что-то есть, но кажется это не сильно влияет. Роль воды в фолдинге малых нуклеиновых кислот
Правда, я не очень понял, как сказанное выше противоречит введению учета гидрофобности/гидрофильности, если оно надо будет?
стэкинг. Если закрутить нитку, то образуются такие же ответвления. Причина этого мне не известна, но похоже имеет место закон сохранения закрученности. Есть ли такой закон для полимерных цепочек, в частности, РНК?
Не вы ли мне задавали вопрос, про то имеет ли отношение к вопросу сворачивания волос, который скручивается :)
Ну — волос это белок, поэтому скручивается тоже по правилам полимерных цепочек. Но вопроса вашего я не понял…
Ну — волос это белок, поэтому скручивается тоже по правилам полимерных цепочек. Но вопроса вашего я не понял…
Как и предыдущий вопрос, этот направлен на попытку перенести задачу на макроуровень, чтобы она стала более наглядной, так как подобное моделирование может оказаться полезным. Если крутить нитку, то могут появиться сдвоенные ответвления, которые сами закручиваются в противоположную сторону, как бы компенсируя закручивание. Немного прояснила википедия: Суперскрученность. Показалось, что более правильная форма скручивания будет более наглядной и простой для понимания и может послужить базой(форма сворачивания по умолчанию) для понимания более сложного сворачивания(модификация базовой формы), например, РНК.
«Но вопроса вашего я не понял…» — если это относилось к вопросу про волос, то там я представлял как рибосома синтезирует белок: точка добавления аминокислоты напомнила точку между ногтями, перемещающуюся по волосу. Возник вопрос насколько далеко можно распространить аналогию: зависит ли форма белка от скорости его синтеза, при каких условиях можно получать стабильную форму волоса на выходе и т.д.
«Но вопроса вашего я не понял…» — если это относилось к вопросу про волос, то там я представлял как рибосома синтезирует белок: точка добавления аминокислоты напомнила точку между ногтями, перемещающуюся по волосу. Возник вопрос насколько далеко можно распространить аналогию: зависит ли форма белка от скорости его синтеза, при каких условиях можно получать стабильную форму волоса на выходе и т.д.
Я вот ни разу не учёный, но смотрите какая шутка получается:
* я так понял, РНК всегда свёрнута одинаково, для всех экземпляров молекулы
* конечная конфигурация молекулы не обязательно будет глобальным энергетическим минимумом ( вы картинку из википедии постили, где это иллюстрируется)
Т.е. по идее молекула может существовать в нескольких стационарных конфигурациях, но получается почему-то только одна. При этом, поправьте меня если это не так, но вроде бы нет экспериментов в которых бралась бы развёрнутая в линию РНК и она бы сама свернулась обратно в свою правильную форму.
Я говорю о том, что, похоже что, моделировать сворачивание целой молекулы смысла нет — она в природе не бывает вот такой вот вытянутой в линию и всё равно в правильную структуру не свернётся. По-моему достаточно очевидно, что молекула начинает сворачиваться по мере того как она строится, вот исходя из этого и нужно плясать.
* я так понял, РНК всегда свёрнута одинаково, для всех экземпляров молекулы
* конечная конфигурация молекулы не обязательно будет глобальным энергетическим минимумом ( вы картинку из википедии постили, где это иллюстрируется)
Т.е. по идее молекула может существовать в нескольких стационарных конфигурациях, но получается почему-то только одна. При этом, поправьте меня если это не так, но вроде бы нет экспериментов в которых бралась бы развёрнутая в линию РНК и она бы сама свернулась обратно в свою правильную форму.
Я говорю о том, что, похоже что, моделировать сворачивание целой молекулы смысла нет — она в природе не бывает вот такой вот вытянутой в линию и всё равно в правильную структуру не свернётся. По-моему достаточно очевидно, что молекула начинает сворачиваться по мере того как она строится, вот исходя из этого и нужно плясать.
Зарегистрируйтесь на Хабре, чтобы оставить комментарий
Биовычисления по сворачиванию. Снова простым языком о полученной модели сворачивания