
Заметил, что статьи получаются довольно большими, и вопросы задаются в разных направлениях. Эта статья была написана для того, чтобы собрать вопросы по установке программы ZINBA в отдельной теме. Итак, для работы с ZINBA нужно знать, как ее установить.
Подробная инсталляция расписана на сайте разработчика на английском языке. Поэтому далее я кратко опишу необходимые этапы на русском. Все шаги выполнялись мною в ОС OpenSUSE 12.1 x64, но должны проходить без эксцессов и на других Linux платформах. Запускаем R (если есть вопросы по настройке, обсудим в комментариях ), выполняем следующие команды:
- system(«wget zinba.googlecode.com/files/zinba_2.01.tar.gz»). Скачивает пакет с сайта разработчика.
- install.packages(c('multicore','doMC','foreach','qvalue','quantreg','R.utils')). Устанавливает дополнительные пакеты. Появится окно со списком сайтов для загрузки, в нем выбрать сайт, с которого надо установить эти пакеты.
- install.packages(«zinba_2.01.tar.gz», repos=NULL). Происходит установка загруженного пакета.
- Идем в раздел «Necessary file downloads», в подразделе «Genome Build» скачиваем один или несколько двухбитных индексов генома, зависит от того с каким из них собираетесь работать.
- В подразделе «Mappability files» загружаем файл, содержащий в названии имя генома и длину ридов, как в вашем FASTA/FASTQ файле. Не забываем разархивировать его в какую-нибудь директорию. В Linux, например, скачать и разархивировать можно так: “wget www.bios.unc.edu/~nrashid/map##.tgz;tar -xzvf map##.tar.gz”, где ## — число, соответствующее длине рида.
- Запускаем R, выполняем последовательно две команды: 1. generateAlignability, для создания индекса выравнивания и 2. basealigncount, для базового подсчета выравниваний; эта команда создаст файл, содержащий точную привязку пиков в несколько итераций.
generateAlignability(
mapdir=, #указать путь к директории, куда был развернут архив «mappability»
outdir=, #путь к директории, в которой будут обрабатываться файлы
athresh=, #количество ошибок, разрешённых при мэппинге ридов на геном
extension=, #средняя предполагаемая длина фрагментов ДНК из эксперимента
twoBitFile=, #путь до файла, двухбитного индекса генома .2bit
)
basealigncount(
inputfile=, #путь до фала с нашими экспериментальными данными, например, после bowtie
outputfile=, # путь к директории, куда выводится результат
extension=, #средняя предполагаемая длина фрагментов ДНК из эксперимента
filetype=, #тип входного файла "bed", "bowtie", or "tagAlign"
twoBitFile=, # путь до файла, двухбитного индекса генома .2bit
)
Первую команду рекомендуется выполнить один раз и в дальнейшем выполнять только, если меняются параметры экспериментов, такие как: количество ошибок и длина фрагментов. Вторую команду следует выполнять от эксперимента к эксперименту, и то только, если нам нужно повышать качество нахождения обогащеных регионов и пиков (так как это требует дополнительной вычислительной нагрузки).
Во время выполнения этих действий я получил ряд ошибок, пообщался со службой технической поддержки. Выяснилось, что я недостаточно хорошо прочитал инструкцию, да и у них не все гладко. Первое, если файл получен с помощью bowtie (обычный не .sam, без параметра –S), то последний столбец в файле надо попросту удалить, столбцов в файле должно быть 7. Также оказалось, что разделителем считается не только табуляция, но и пробел, поэтому следите, чтобы поля не содержали пробелы. Это актуально для версии 2.01, обещали в будущем исправить.
Сама команда нахождения обогащённых регионов описана ниже, выполняется долго и съедает все ресурсы. Я выполнял её на экспериментальных данных с 33.000.000 ридов, на 4х CPU (не путать с камнем, просто 4ре threads, один Core i7), 8Gb RAM, правда в виртуальной машине VirtualBOX, вычисления заняли 8 часов. При наличии всего 8Gb RAM ставить больше threads не рекомендую, программа будет больше работать с файлом подкачки, нежели с данными.
zinba(
refinepeaks=, #повышать качество пиков (очищать регион)? 1 - если да, 0 - если нет
seq=, #путь к файлу с экспериментальными данными
input=, #путь к файлу с экспериментальными данными, которые найдены на геноме, если файл доступен. По умолчанию равно ‘none’
filetype=, #формат файлов: 'bed', 'bowtie', или 'tagAlign'
threshold=, #уровень, при каком значении p value считать данные значимыми. По умолчанию менее, чем 0.05
align=, #путь к директории, указанный как outdir в функции generateAlignability
numProc=, #количество CPU должно быть меньше, чем максимально доступное количество (у Core i7 их 8, ставим 7 или меньше, по умолчанию 1)
twoBit=, #путь до файла, двухбитного индекса генома .2bit
outfile=, #путь вместе с префиксом для файла
extension=, #средняя предполагаемая длина фрагментов ДНК из эксперимента
###################
ОПЦИОНАЛЬНЫЕ ПАРАМЕТРЫ
###################
basecountfile=, #путь к basecount файлу, если параметр refinepeaks установлен в 1
broad=, #граничные значения, TRUE или FALSE (по умолчанию)
printFullOut=, #сохранять в файлы промежуточные вычисления: 1, если да (требуется много места); 0, если нет (по умолчанию)
)
Мы подготовили пакет к работе, теперь самое время запустить что-нибудь для его проверки. Разработчик предлагает набор файлов для теста: http://www.unc.edu/~nur2/zinbaweb/test_data.tgz. В командой строчке последовательно выполняем следующие действия.
#Открываем терминал, скачиваем в текущую директорию файлы с сайта.
wget http://hgdownload.cse.ucsc.edu/goldenPath/hg19/bigZips/hg19.2bit
wget http://www.bios.unc.edu/~nrashid/map36.tgz
wget http://www.unc.edu/~nur2/zinbaweb/test_data.tgz
#Распаковываем скаченные архивы
tar -xzvf map36.tgz
tar -xzvf test_data.tgz
#Создаем директорию для данных, например, align_athresh1_extension200
mkdir align_athresh1_extension200
#Запускаем R.
R
#в R выполняем последовательно следующие команды
library(zinba)
generateAlignability(
mapdir='map36/',
outdir='align_athresh1_extension200/',
athresh=1,
extension=200,
twoBitFile='hg18.2bit'
)
basealigncount(
inputfile='data/ctcfGM12878rep3chr22.taf',
outputfile='data/ctcfGM12878rep3chr22.basecount',
extension=200,
filetype='tagAlign',
twoBitFile='hg18.2bit' )
zinba(
align='align_athresh1_extension200/',
numProc=4,
seq='data/ctcfGM12878rep3chr22.taf',
basecountfile='data/ctcfGM12878rep3chr22.basecount',
filetype="tagAlign",
outfile="data/ctcf",
twoBit="hg18.2bit",
extension=200,
printFullOut=1,
refinepeaks=1,
broad=F,
input='data/inputGM12878rep3chr22.taf'
)
Я работал с другими данными, взятыми с сайта NIH www.ncbi.nlm.nih.gov/geo/roadmap/epigenomics, ftp-trace.ncbi.nlm.nih.gov/sra/sra-instant/reads/ByExp/sra/SRX/SRX040/SRX040388 (файл SRR079566.sra). Ниже приведено изображение, полученное с использованием этих данных. Похожие графики могут быть получены и при других исходных данных. С помощью красной линии я разделил картинку на две части, сверху расположены экспериментальные данные, внизу стандартная аннотация (гены, старт сайты, интроны, эксоны и т.д.). Я загрузил не только полученный bedgraph из программы ZINBA, но и исходные данные (bedgraph по исходным данным получен с помощью программы, описанной в статье habrahabr.ru/blogs/bioinformatics/137082). Для примера я выделил зелеными квадратами несколько пиков, которые были в исходных данных, и те области обогащения, которые нашла ZINBA. Отрезки, полученные из ZINBA, содержат утолщения. Эти утолщения подсчитываются при заданном параметре refignpeaks=1 и должны соответствовать наиболее обогащенной части региона.
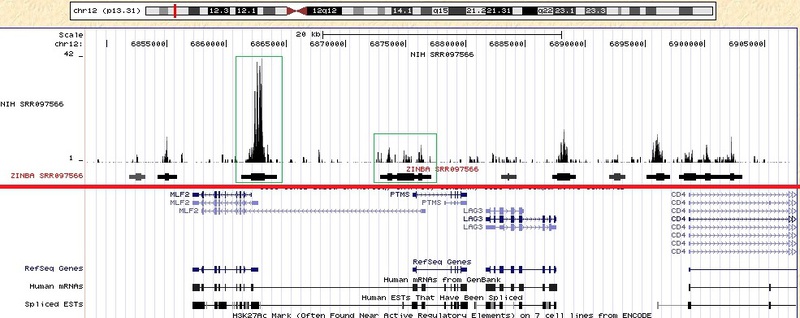
Насколько правильно и достоверно обработаны данные, можно будет понять только после досконального изучения алгоритма и сравнения с результатом работ других программ.
Review is prepared by Andrey Kartashov, Cincinnati, OH, porter@porter.st







