Не, ну какая рукотворность? Что за бред? Думал я, когда впервые услышал гипотезу о том, что Ковид-19 вызван то ли лабораторной утечкой, то ли вообще целенаправленной биоатакой. И каждый раз просто отмахивался от этих домыслов, когда они в очередной раз доплывали до меня в бурном потоке коронавирусного инфошума. Ну подумаешь, есть в Ухане институт вирусологии, мало ли.
В какой-то момент отмахиваться уже пришлось аргументированно, потому что сторонники рукотворности начали обосновывать свои тезисы о возможной искусственной природе вируса доводами из молекулярной биологии, и тут уже хотелось в пух и прах разбить их конспирологию холодными научными фактами. Уж если не как авторы статьи в Nature (казалось мне), то хотя бы как уважаемый мной Панчин.
И вот тут, в погоне за доводами против рукотворности вируса, меня и заразил вирус сомнений. В чём, собственно, причина сомнений? В том, что чем глубже погружаешься в деятельность коронавирусологов за последние 15–20 лет, тем лучше понимаешь, что создание ровно таких химер как CoV2 у них было обыденным делом. А CoV2 — это очевидная химера, основанная на летучемышином штамме RaTG13, у которого в шиповидном белке место связывания с рецептором (RBM) заменено с летучемышиного на панголиний, и вдобавок врезан особый участок из 4-х аминокислот, создавший furin cleavage site, который, как ранее выяснили вирусологи, значительно расширяет «репертуар» вируса в плане того, в чьи клетки он может проникать. Скорее всего, именно благодаря этому новому фуриновому сайту, новый мутант и сумел перескочить с исходных носителей на людей.
С учётом тех высот, которых сегодня достигла генная инженерия, синтетически собрать CoV2 по вышеописанной методике не составило бы труда даже начинающему специалисту. Ведь вирусологи, включая руководителя коронавирусного направления в Уханьском институте вирусологии Ши Чжэнли, такими вещами уже неоднократно занимались — как заменой RBM у одного вида вируса на RBM из другого (вот работа группы Ши Чжэнли от 2007 года), так и добавлением нового фуринового сайта, способного дать специфичному к одному виду животных коронавирусу возможность начать использовать рецептор ACE2 других видов.
Минутка заботы от НЛО
В мире официально объявлена пандемия COVID-19 — потенциально тяжёлой острой респираторной инфекции, вызываемой коронавирусом SARS-CoV-2 (2019-nCoV). На Хабре много информации по этой теме — всегда помните о том, что она может быть как достоверной/полезной, так и наоборот.
Мы призываем вас критично относиться к любой публикуемой информации
Официальные источники
- Cайт Министерства здравоохранения РФ
- Cайт Роспотребнадзора
- Сайт ВОЗ (англ)
- Сайт ВОЗ
- Сайты и официальные группы оперативных штабов в регионах
Если вы проживаете не в России, обратитесь к аналогичным сайтам вашей страны.
Мойте руки, берегите близких, по возможности оставайтесь дома и работайте удалённо.
Читать публикации про: коронавирус | удалённую работу
Ши Чжэнли в своей лаборатории в Уханьском институте вирусологии
Но прежде чем разводить тут конспирологию, давайте сначала окунёмся в биологию.
Биология
Итак, начнём от печки. Что за фуриновый сайт, что за RBM, что вообще за шиповидный белок? На самом деле, если продраться сквозь дебри терминологии, то концептуально всё просто. Вот, например, шиповидный белок — это та самая торчащая из вирусной частицы штука (S protein), за которую эти вирусы и “короновали”:
Именно с помощью этих белков вирион цепляется за рецептор клетки-жертвы (ACE2 в нашем случае), чтобы затем проникнуть внутрь. Поэтому можно сказать, что это самая важная часть вируса, ведь именно она определяет каких животных тот может поражать, а каких нет — ACE2 рецепторы у разных видов немного структурно отличаются. При этом из всего огромного по вирусным меркам 30-килобазного генома, ген этого белка составляет лишь 12–13%, то есть около 1300 аминокислот. Вот так структурирован шиповидный белок у CoV2 и его близких родственников:
Как видно из рисунка выше, S белок состоит из двух субъединиц: S1 и S2. Именно S1 взаимодействует с рецептором ACE2, и то место, которым он это делает, называется Receptor Binding Domain (RBD), а область непосредственного контакта, святая святых, называется Receptor Binding Motif (RBM). Вот красивая иллюстрация из не менее красивой работы:
Общая структура RBD CoV2, связанного с ACE2.
(а) Общая топология мономера шиповидного белка 2019-nCov. NTD, N-терминальный домен. RBD, рецептор-связывающий домен. RBM, рецептор-связывающий мотив. SD1, поддомен 1. SD2, поддомен 2. FP, пептид слияния. HR1, гептадный повтор 1. HR2, гептадный повтор 2. TM, трансмембранная область. IC, внутриклеточный домен.
(б) Последовательность и вторичные структуры RBD 2019-nCov. RBM окрашен в красный цвет.
(с) Общая структура RBD 2019-nCov, связанного с ACE2. ACE2 окрашен в зеленый цвет. Ядро RBD 2019-nCoV окрашено в голубой цвет, а RBM — в красный. Дисульфидные связи в RBD 2019-nCov показаны в виде линии и обозначены желтыми стрелками. N-концевая спираль ACE2, ответственная за связывание, помечена.
Исходный текстOverall structure of 2019-nCoV RBD bound with ACE2.
(a) Overall topology of 2019-nCoV spike monomer. NTD, N-terminal domain. RBD, receptor-binding domain. RBM, receptor-binding motif. SD1, subdomain 1. SD2, subdomain 2. FP, fusion peptide. HR1, heptad repeat 1. HR2, heptad repeat 2. TM, transmembrane region. IC, intracellular domain.
(b) Sequence and secondary structures of 2019-nCoV RBD. The RBM is colored red.
(с) Overall structure of 2019-nCoV RBD bound with ACE2. ACE2 is colored green. 2019-nCoV RBD core is colored cyan and RBM is colored red. Disulfide bonds in the 2019-nCoV RBD are shown as stick and indicated by yellow arrows. The N-terminal helix of ACE2 responsible for binding is labeled.
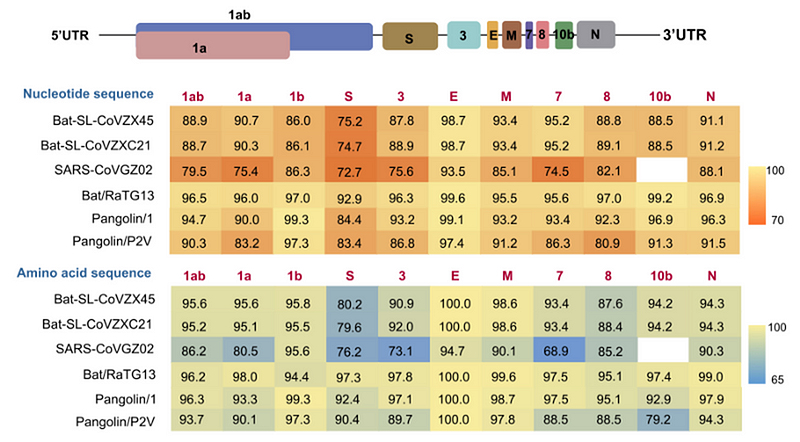
Так вот. Когда геном CoV2 только расшифровали, то сначала не было известно каких-то прям близкородственных ему штаммов. Но уже 23 января 2020 года Ши Чжэнли выпустила работу, в которой объявила, что CoV2 на 96% совпадает со штаммом RaTG13, который её лаборатория в 2013 году выделила из Юньнаньских летучих мышей. Правда, вне её лаборатории до января 2020 года об этом штамме не было известно никому.
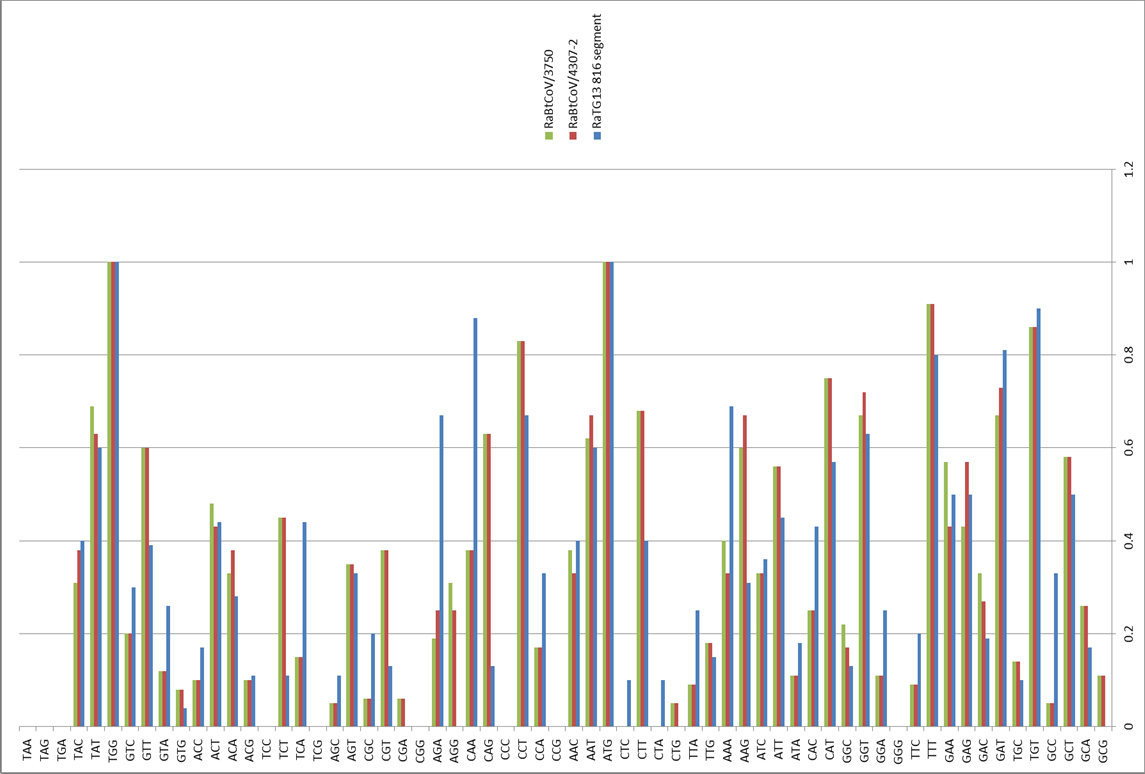
Было сразу понятно, что RaTG13 — мальчик особенный. Взгляните на график:
Это график схожести CoV2 с другими известными штаммами. Чем выше кривая, тем выше процент совпадения нуклеотидов. Как видим, в районе гена того самого шиповидного белка (S) только RaTG13 более-менее близок к CoV2, а все остальные штаммы в этом месте уходят в пике — как штаммы вирусов из других летучих мышей, так и первый SARS-CoV (красная кривая). Но тут пока нет ничего подозрительного — мало ли ещё неизвестных науке штаммов таят в себе неизведанные Юньнаньские пещеры? Ну да, не очень понятно как именно вирус оттуда добрался до Уханя, но чего не бывает.
Панголины
Далее на сцене появляются панголины: в феврале другая группа китайских учёных обнаружила в своих закромах штамм панголиньего коронавируса, который, хоть в целом и хуже чем RaTG13 был похож на CoV2 (на 90%), в том самом RBM шиповидного белка был почти идентичен — отличался лишь на 1 аминокислоту (см. два верхних сиквенса, точки означают совпадение с первым сиквенсом):

При этом в первой четверти S белка панголиний штамм на CoV2 не похож, а последовательность после RBM у всех троих штаммов (CoV2, Pangolin, RaTG13) более-менее совпадает. Но вот сам RBM у RaTG13 весьма отличается от CoV2, что видно по крутому провалу зелёного графика RaTG13 по сравнению с красным графиком CoV2 в районе RBM (вертикальная розовая полоса) на следующем графике:
Эту разницу подтверждает и филогенетический анализ этих трёх областей, выделенных на графике выше — по RBM штамм панголина ближе к CoV2, чем RaTG13, а вот слева и справа от RBM к CoV2 ближе именно RaTG13. То есть налицо явная рекомбинация, о чем упоминают и сами авторы, и некоторые другие работы.
Кстати, откуда взялись у исследователей эти самые панголины? А вот отсюда:

Их конфисковала у контрабандистов китайская таможня и передала в реабилитационный центр в Гуандуне, где они скончались при выраженной коронавирусной симптоматике. Это, конечно, не могло не заинтересовать местных вирусологов, которые и выделили у них различный биоматериал:
Панголины, использованные в исследовании, были конфискованы таможней и Департаментом лесного хозяйства провинции Гуандун в марте-декабре 2019 года. В их число входят четыре китайских панголина (Manis pentadactyla) и 25 малайских панголинов (Manis javanica). Эти животные были отправлены в центр спасения диких животных; в основном они были неактивны и задыхались, и в конечном итоге умерли, несмотря на обширные усилия по их спасению. Образцы тканей были взяты из легких, лимфатических узлов, печени, селезенки, мышц, почек и других тканей у только что умерших панголинов, для гистопатологических и вирусологических исследований.Кстати, и не только местных, потому что другие китайские исследователи (гонконгские в данном случае) тоже получали образцы конфискованных панголинов и в феврале 2020 тоже выпустили схожую работу, отметив явные признаки рекомбинации в шиповидном белке CoV2:
Исходный текстPangolins used in the study were confiscated by Customs and Department of Forestry of Guangdong Province in March-December 2019. They include four Chinese pangolins (Manis pentadactyla) and 25 Malayan pangolins (Manis javanica). These animals were sent to the wildlife rescue center, and were mostly inactive and sobbing, and eventually died in custody despite exhausting rescue efforts. Tissue samples were taken from the lung, lymph nodes, liver, spleen, muscle, kidney, and other tissues from pangolins that had just died for histopathological and virological examinations.
Мы получили образцы замороженных тканей (легких, кишечника, крови), которые были взяты у 18 малайских панголинов (Manis javanica) в течение августа 2017 года — января 2018 года. Эти панголины были получены в ходе операций по борьбе с контрабандой таможней Гуанси. Поразительно, что высокопроизводительное секвенирование их РНК выявило присутствие коронавирусов в шести (два легких, два кишечника, одна смесь легких и кишечника, одна кровь) из 43 образцов.Кстати, авторы этой статьи тоже выделили явную филогенетическую мозаичность шиповидного белка CoV2:
…
Более заметным, однако, было наблюдение предполагаемых сигналов рекомбинации между коронавирусами панголинов, коронавирусами летучих мышей RaTG13 и 2019-CoV2 человека (Рис. 1c, d). В частности, 2019-CoV2 демонстрирует очень высокое сходство последовательностей с коронавирусами панголина из провинции Гуандун в рецептор-связывающем домене (RBD; сходство аминокислот 97,4%; обозначено красной стрелкой на рис. 1c и рис. 2a), хотя в оставшейся части вирусного генома 2019-CoV2 ближе всего к RaTG13. В то же время RaTG13 и 2019-CoV2 имеют только 89,2% сходства аминокислот в RBD. Коронавирусы панголинов из Гуандуна и 2019-CoV2 содержат идентичные аминокислоты в пяти критических остатках RBD, тогда как RaTG13 имеет только одну общую аминокислоту с 2019-CoV2 из этих пяти (остаток 442, нумерация SARS-CoV человека).
Исходный текстWe received frozen tissue (lungs, intestine, blood) samples that were collected from 18 Malayan pangolins (Manis javanica) during August 2017-January 2018. These pangolins were obtained during the anti-smuggling operations by Guangxi Customs. Strikingly, high-throughput sequencing of their RNA revealed the presence of coronaviruses in six (two lung, two intestine, one lung-intestine mix, one blood) of 43 samples. With the sequence read data, and by filling gaps with amplicon sequencing, we were able to obtain six full or nearly full genome sequences — denoted GX/P1E, GX/P2V, GX/P3B, GX/P4L, GX/P5E and GX/P5L — that fall into the 2019-CoV2 lineage (within the genus Betacoronavirus) in a phylogenetic analysis (Figure 1a).
…
More notable, however, was the observation of putative recombination signals between the pangolins coronaviruses, bat coronaviruses RaTG13, and human 2019-CoV2 (Figure 1c, d). In particular, 2019-CoV2 exhibits very high sequence similarity to the Guangdong pangolin coronaviruses in the receptor-binding domain (RBD; 97.4% amino acid similarity; indicated by red arrow in Figure 1c and Figure 2a), even though it is most closely related to bat coronavirus RaTG13 in the remainder of the viral genome. Bat CoV RaTG and the human 2019-CoV2 have only 89.2% amino acid similarity in RBD. Indeed, the Guangdong pangolin coronaviruses and 2019-CoV2 possess identical amino acids at the five critical residues of the RBD, whereas RaTG13 only shares one amino acid with 2019-CoV2 (residue 442, human SARS-CoV numbering).
Интересно, что филогенетический анализ только синонимичных сайтов в RBD показал, что филогенетическое положение гуандунского панголина согласуется с положением по остальной части вирусного генома, а именно, что он не является ближайшим родственником 2019-CoV2 (Figure 2b). Следовательно, возможно, что сходство аминокислот в RBD коронавирусов панголина и 2019-CoV2 обусловлено селективно-опосредованной конвергентной эволюцией, а не рекомбинацией, хотя на имеющихся данных трудно выбрать между этими сценариями.В переводе с научного, слова авторов означают, что если проанализировать весь RBD трёх штаммов, отбросив явные различия (несинонимичные замены) между ними, которые, в основном, приходятся на RBM (который, напомню, идентичен между CoV2 и Pangolin), и построить филогенетическое древо по синонимичным заменам, то CoV2 таки ближе к RaTG13, а не к панголиньему штамму. Что довольно странно в свете того, что у панголиньего с CoV2 идентичный RBM (то есть отрезок внутри RBD).
Исходный текстInterestingly, a phylogenetic analysis of synonymous sites alone in the RBD revealed that the phylogenetic position of the Guangdong pangolin is consistent with that in the remainder of the viral genome, rather than being the closest relative of 2019-CoV2 (Figure 2b). Hence, it is possible that the amino acid similarity between the RBD of the Guangdong pangolin coronaviruses and 2019-CoV2 is due to selectively-mediated convergent evolution rather than recombination, although it is difficult to choose between these scenarios on current data.
Далее авторы теоретизируют, что это может быть следствием конвергентной эволюции, то есть, иначе говоря, что штаммы CoV2 и панголинов пришли к идентичному RBM каждый своей дорогой, а не через совместную рекомбинацию общих предков. Потому что действительно уж очень странная должна была произойти рекомбинация — как будто кто-то просто взял кусок RBM из панголиньего штамма и заменил им RBM в RaTG13. А это уже смахивает не на эволюцию, а на, прости Дарвин, Intelligent Design.
Генеалогия коронованных особ
Для того чтобы получше понять происхождение CoV2, давайте ещё раз взглянем на последовательности шиповидного белка у нашей троицы: CoV2, RaTG13 и панголина — сравним попарную разницу между ними (точками отмечены идентичные аминокислоты, красные буквы показывают разницу, а прочерки обозначают удалённые/добавленные аминокислоты):

Невооружённым глазом видно, что в первой четверти сиквенса панголиний штамм далёк от CoV2 и RaTG13. Ну а RaTG13, если бы не участок в районе RBM (красный прямоугольник), был бы ооочень близок к CoV2. Но, как я уже говорил, тот самый участок у CoV2 ближе всего к панголиньему штамму.
Кстати, а что там у других панголиньих штаммов? Давайте посмотрим. Ведь пока что мы анализировали только вирус, выделенный из панголинов, конфискованных таможней в 2019 году. А ещё была партия панголинов, конфискованных в 2017-ом, и у них тоже был выделен похожий штамм. Если сравнить RaTG13 с геномами вирусов из панголинов 2017 и 2019 годов, то тут тоже всё интересно:

В первой четверти S белка панголиньи штаммы от 2017 года ближе к RaTG13 (и CoV2) чем их панголиний собрат от 2019-го (MP789). При этом, у всех трёх явный недавний общий предок в областях, выделенных зелёными прямоугольниками, и в этих областях RaTG13 и панголин-19 (MP789) к нему ближе, чем панголин-17, так как у него с RaTG13 несколько общих мутаций (обведены красными и синими эллипсами), которых не наблюдается у легенды-17. При этом RBM у всех трёх разный, и разный примерно в одинаковой пропорции, и в схожих местах.
Возможно, уже после того как предки RaTG13 и MP789 разошлись, у MP789 произошла замена большого участка в первой четверти S белка (которой не произошло у RaTG13 и панголина-17), а остальная часть S белка у всех троих осталась общей. А потом пути генофондов RaTG13 и MP789 сошлись опять и в порыве страсти породили CoV2. Также вполне возможно, что предок RaTG13 является результатом рекомбинации предков панголиньих штаммов.
Ещё любопытно видеть довольно уникальную одинаковую мутацию (QTQTNS) у RaTG13 и панголина-19 прямо перед тем местом, где у CoV2 находится новый furin cleavage site, возникший из-за врезки 4-х новых аминокислот в этом месте (PRRA). Если посмотреть на нуклеотидную последовательность вокруг этой одинаковой мутации, то видно, что RaTG13 и CoV2 по ней ближе друг к другу, чем к панголину-19, так как успели накопить несколько общих мутаций (выделены синим):

Кстати, с Orf1ab у CoV2 тоже филогенетическая чехарда: 1a ближе к RaTG13, а вот 1b — к панголину-19:
То есть получается, что предок CoV2 согрешил с общим предком панголина-19, как минимум, дважды? Впервые — когда он (вместе с общим предком с RaTG13) унаследовал Orf1ab и вторую часть шиповидного белка с QTQTNS мутацией, а вторично — когда приобрёл 1b вместе с RBM, уже отличающимся от RaTG13-шных. Нет, конечно, такое возможно и само по себе не особо примечательно — ведь эти вирусы мутируют и рекомбинируют постоянно. Другой вопрос, где именно летучемышиные и панголиньи вирусы могут друг с другом встречаться для таких оргий — в горных пещерах, “мокрых рынках”, приютах для конфискованных животных, или даже в лабораториях. Но давайте повременим с этими вопросами. Сначала рассмотрим самый бросающийся в глаза аспект нового вируса — врезку из 4-х аминокислот, превратившую его в супер-убийцу.
Врежу аккуратно, но сильно
Совершенно невозможно игнорировать эту врезку PRRA между S1 и S2. Как заноза торчит она в геноме нашего нового CoV2. Врезка эта не простая, а золотая. Она создаёт тот самый furin cleavage site, о котором я упомянул в самом начале. Что это за зверь? Попытаюсь вкратце объяснить. Помните структуру нашего шиповидного белка? Вот наглядная диаграмма:
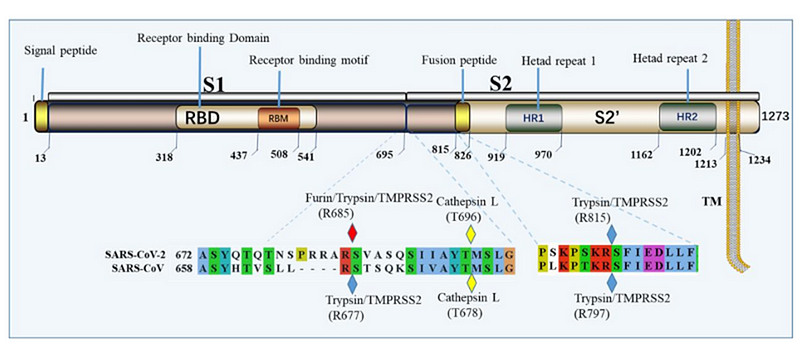
Белок состоит из двух частей, S1 и S2, из которых S1 отвечает за первичный контакт с рецептором (тот самый Receptor Binding Domain/Motif), а S2 отвечает уже за слияние с клеточной мембраной и проникновение в клетку. Запускает процесс слияния отмеченный жёлтым fusion peptide, но для того чтобы он начал своё грязное дело, кто-то должен разрезать S белок в одном из сайтов, выделенных ромбиками на диаграмме выше. Своих таких “резальщиков” у вируса нет (есть другие, но это к делу не относится), поэтому он полагается на различные протеазы своих жертв, благо таких протеаз, как можно понять по обилию цветов тех самых ромбиков, существует несколько видов. Но не все они равны, и не во всех типах клеток есть нужные вирусу протеазы. А фурин как раз один из самых эффективных, да ещё и обитает он не только на поверхности клеток, но и внутри. Нагляднее всего опасность нового фуринового сайта демонстрирует разница между CoV2 и его “предтечей” SARS-CoV:
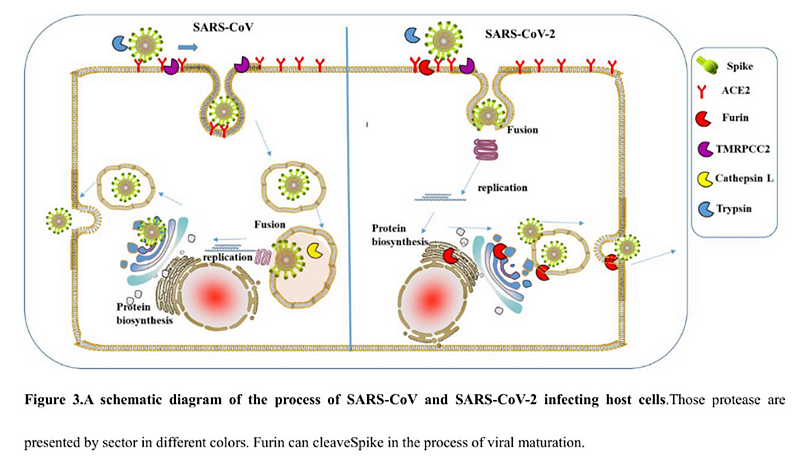
Как видно из диаграммы, в случае CoV2, благодаря фуриновому сайту, разрезать его S белок вне клетки могут не два, а три класса протеаз (три разноцветных пакмэна). Но, быть может, самая важная разница состоит в том, что фурин присутствует и внутри клетки, поэтому он может разрезать S белок сразу после сборки вириона, тем самым повышая способность новых вирионов сливаться с другими клетками — так сказать, не отходя от кассы.
Кстати, существует вероятность, что именно новый фуриновый сайт играет важную роль в выраженной возрастозависимой морбидности и летальности CoV2:
По неизвестным причинам, пациенты с гипертонией, диабетом, ишемической болезнью сердца, цереброваскулярными заболеваниями, хронической обструктивной болезнью легких и почечной дисфункцией имеют худшие клинические исходы при инфицировании SARS-CoV-2. Целью данного обзора является обобщение доказательств существования повышенного плазмин(оген)а у пациентов с COVID-19 с этими коморбидными состояниями. Плазмин и другие протеазы могут расщеплять новый фуриновый сайт в шиповидном белке SARS-CoV-2 внеклеточно, что увеличивает его инфекционность и вирулентность.Ах, да, разрезает фурин белки в строго определённых местах, а именно после последовательности RxxR (то есть Arg-X-X-Arg, где X может быть любой аминокислотой). При этом если на втором или третьем месте тоже аргинин (то есть RRxR или RxRR), то эффективность расщепления этого сайта существенно возрастает.
Исходный текстPatients with hypertension, diabetes, coronary heart disease, cerebrovascular illness, chronic obstructive pulmonary disease, and kidney dysfunction have worse clinical outcomes when infected with SARS-CoV-2, for unknown reasons. The purpose of this review is to summarize the evidence for the existence of elevated plasmin(ogen) in COVID-19 patients with these comorbid conditions. Plasmin, and other proteases, may cleave a newly inserted furin site in the S protein of SARS-CoV-2, extracellularly, which increases its infectivity and virulence.
Поэтому появление нового furin cleavage site специалистами было замечено сразу. Ещё бы! Ведь такого сайта нет ни у кого из ближайших или даже дальних родственников Cov2 — те коронавирусы, у которых он есть, лишь на 40% близки к Cov2 по геному:
Было обнаружено, что все шиповидные белки с гомологией белку SARS-CoV-2, превышающей 40%, не имели сайта расщепления фурином (рисунок 1, таблица 1), включая Bat-CoV RaTG13 и SARS-CoV (с идентичностью последовательности как 97,4 % и 78,6% соответственно). Фуриновый сайт «RRAR» в SARS-CoV-2 является уникальным в своем семействе, благодаря уникальной вставке «PRRA». Этот сайт в SARS-CoV-2 вряд ли мог эволюционировать из MERS, HCoV-HKU1 и т.д. Из имеющихся в настоящее время последовательностей в базах данных нам трудно найти источник. Возможно, есть еще много эволюционных промежуточных последовательностей, ожидающих открытия.Вот наглядная иллюстрация из той же статьи, что и вышеприведённая цитата (розовым отмечены коронавирусы с фуриновым сайтом, на 10 часах приведены 3 разных штамма Cov2):
Исходный текстIt was found that all Spike with a SARS-CoV-2 Spike sequence homology greater than 40% did not have a furin cleavage site (Figure 1, Table 1), including Bat-CoV RaTG13 and SARS-CoV (with sequence identity as 97.4% and 78.6%, respectively). The furin cleavage site “RRAR” in SARS-CoV-2 is unique in its family, rendering by its unique insert of “PRRA”. The furin cleavage site of SARS-CoV-2 is unlikely to have evolved from MERS, HCoV-HKU1, and so on. From the currently available sequences in databases, it is difficult for us to find the source. Perhaps there are still many evolutionary intermediate sequences waiting to be discovered.
Ближайший родственник с фуриновым сайтом — это штамм HKU5, выделенный командой Ши Чжэнли в 2014 году в Гуанчжоу из летучих мышей рода Pipistrellus (добавлен в GenBank в 2018-ом). Но родственник он весьма дальний — их шиповидные белки совпадают лишь на 36%.
В общем, учёные в замешательстве. Откуда взялась эта вставка из 12 нуклеотидов? Может ли она быть рукотворной? Вполне. Ведь такими вставками вирусологи занимались многократно, причём издавна. Например, американцы вставляли RRSRR в шиповидный белок первого SARS-CoV ещё в 2006-ом:
Чтобы исследовать, может ли протеолитическое расщепление в месте последовательсти основных аминокислотных остатков способствовать клеточной активности слияния, мы мутировали шиповидный белок SARS-CoV, чтобы создать сайт узнавания фурином (RRSRR) в одном из двух мест.
Исходный текстTo investigate whether proteolytic cleavage at the basic amino acid residues, were it to occur, might facilitate cell–cell fusion activity, we mutated the wild-type SARS-CoV glycoprotein to construct a prototypic furin recognition site (RRSRR) at either position.

А японцы вставили такой сайт (RRKR) в белок SARS-CoV в 2008 году, правда немного ниже по течению:
В том же 2008-ом году их голландские коллеги тоже изучали эти протеазные сайты у SARS-CoV и сравнивали их с мышиным коронавирусом MHV, у которого этот сайт есть (SRRAHR|SV), причём похожий на сайт нашего CoV2 (SPRRAR|SV):
В 2009 году другая американская группа тоже решила поупражняться над “улучшением” SARS-CoV и, не изменяя американской традиции не мелочиться с аргининами, они вставили RRSRR:
Чтобы исследовать потенциальное использование SARS-CoV S1-S2 и S2 ‘положений в качестве сайтов для протеолитического расщепления, мы сначала ввели сайты распознавания расщепления фурином в этих местах, сделав следующие мутации 664-SLLRSTSQSI — SLLRRSRRSI-671 (S1-S2) и 792-LKPTKRSF-LKRTKRSF-799 (S2 ‘).
Исходный текстTo examine the potential use of the SARS-CoV S1–S2 and S2? positions as sites for proteolytic cleavage, we first introduced furin cleavage recognition sites at these locations by making the following mutations 664-SLLRSTSQSI — SLLRRSRRSI-671 (S1–S2) and 792-LKPTKRSF — LKRTKRSF-799 (S2?).
Пекин-2019
Но самой недавней подобной работой, которую я видел, была работа октября 2019 года учёных из Пекина, где новый фуриновый сайт RRKR вставили не в какой-то псевдовирус, а в самый настоящий куриный коронавирус, infectious bronchitis virus (IBV):
Кстати, интересно упоминание авторов, что добавление фуринового сайта позволяет вирусу-мутанту поражать нервные клетки. Быть может, именно фуриновый сайт CoV2 является причиной того, что некоторые пациенты с CoV2 демонстрируют неврологическую симптоматику, включая потерю обоняния:
Мутация сайта S2' шиповидного белка рекомбинантного вируса генотипа QX приводит к более высокой патогенности, выраженным нервным симптомам и нейротропизму по сравнению с цыплятами, инфицированными вурусом дикого типа IBV (WT-IBV). В этом исследовании мы представляем доказательства того, что рекомбинантный IBV с мутантным сайтом S2 (фуриновый сайт-S2) приводит к более высокой смертности. Заражение мутантом IBV вызывает тяжелый энцефалит и разрушает гематоэнцефалический барьер.При этом у многих коронавирусов фуриновые сайты, конечно же, встречаются в природе, и они весьма разнообразны. Да и появляться они могут и в результате случайных мутаций. Именно это произошло в случае с MERS-ом, о чем в 2015 нам поведал международный коллектив авторов, среди которых были и Ши Чжэнли, и Ральф Барик — две звезды синтетической коронавирусологии. О них мы ещё неоднократно вспомним, а пока пару слов о той статье. Собственно в ней авторы показали две ключевые мутации, которые позволили MERS перескочить с летучих мышей на человека. И одна из этих мутаций привела к возникновению фуринового сайта. Правда это была не врезка новых аминокислот, а мутации ранее существующих (отмеченных красным ниже):
…
Таким образом, наши результаты демонстрируют, что фуриновый сайт перед FP в шиповидном белке является важным сайтом для CoV, модулирующим проникновение, слияние клеток с вирусом, адаптацию к клетке-хозяину, клеточный тропизм и патогенность, но не антигенность.
Исходный текстMutation of the S2' site of QX genotype (QX-type) spike protein (S) in a recombinant virus background results in higher pathogenicity, pronounced neural symptoms and neurotropism when compared with conditions in wild-type IBV (WT-IBV) infected chickens. In this study, we present evidence suggesting that recombinant IBV with a mutant S2' site (furin-S2' site) leads to higher mortality. Infection with mutant IBV induces severe encephalitis and breaks the blood–brain barrier.
…
In summary, our results demonstrate that the furin cleavage site upstream of the FP in S protein is an important site for CoV, modulating entry, cell–virus fusion, adaptation to its host cell, cell tropism and pathogenicity, but not antigenicity.
Авторы не просто показали, а привязали эти мутации обратно к исходному летучемышиному шиповидному белку, создав в нём те же самые мутации (то есть, и фуриновый сайт), и показав, что это придаёт ему способность заражать человеческие клетки:
Чтобы оценить потенциальные генетические изменения, необходимые для HKU4 для заражения клеток человека, мы изменили шиповидный белок HKU4, стремясь повысить его способность опосредовать проникновение вируса в клетки человека. С этой целью мы ввели две одиночные мутации, S746R и N762A, в белок HKU4.Кстати, то как они это сделали может людей далёких от современных биотехнологий напугать само по себе — потому что авторы вставили этот коронавирусный шиповидный белок в инактивированный ретровирус (ВИЧ):
…
Следовательно, реинжинирированные мотивы hPPC и hECP позволили активировать спайк HKU4 человеческими эндогенными протеазами и тем самым позволили псевдовирусам HKU4 обойти необходимость проникновения экзогенных протеаз в клетки человека. Эти результаты показывают, что спайку HKU4 нужны только две одиночные мутации на границе S1/S2, чтобы получить полную способность опосредовать проникновение вируса в клетки человека.
Исходный текстTo evaluate the potential genetic changes required for HKU4 to infect human cells, we reengineered HKU4 spike, aiming to build its capacity to mediate viral entry into human cells. To this end, we introduced two single mutations, S746R and N762A, into HKU4 spike. The S746R mutation was expected to restore the hPPC motif in HKU4 spike, whereas the N762A mutation likely disrupted the potential N-linked glycosylation site in the hECP motif in HKU4 spike.
…
Therefore, the reengineered hPPC and hECP motifs enabled HKU4 spike to be activated by human endogenous proteases and thereby allowed HKU4 pseudoviruses to bypass the need for exogenous proteases to enter human cells. These results reveal that HKU4 spike needs only two single mutations at the S1/S2 boundary to gain the full capacity to mediate viral entry into human cells.
Вкратце, псевдотипированные ретровирусы MERS-CoV-spike, экспрессирующие репортерный ген люциферазы, были получены путем котрансфекции клеток HEK293T плазмидой, несущей геном ВИЧ-1, дефектный по Env, экспрессирующий люциферазу (pNL4–3.luc.RE-) и плазмидой, кодирующей MERS-CoV шиповидный белок.Быть может, именно это подвигло индийских исследователей на поиск схожих у ВИЧ и CoV2 последовательностей в геноме (но их препринт быстро раскритиковали за неудачную методологию и ошибочные выводы). На самом деле такие псевдовирусы специалисты используют регулярно, и вообще, не стоит огульно бояться ретровирусов как класс — их подвид лентивирусы используется для генной терапии уже много лет.
Исходный текстBriefly, MERS-CoV-spike-pseudotyped retroviruses expressing a luciferase reporter gene were prepared by cotransfecting HEK293T cells with a plasmid carrying Env-defective, luciferase-expressing HIV-1 genome (pNL4–3.luc.R-E-) and a plasmid encoding MERS-CoV spike protein.
Откуда взялся RaTG13
Вообще, RaTG13 — феноменальный штамм. Странно, что все эти годы группа Ши Чжэнли о нём молчала. Ведь он совсем не похож на остальных своих собратьев, особенно в последовательности шиповидного белка, а это, напомню, именно то место, которое определяет к каким типам клеток (и каких видов) данный вирус сможет цепляться. Вот график схожести генома CoV2 в сравнении с другими летучемышиными коронавирусами (панель B):
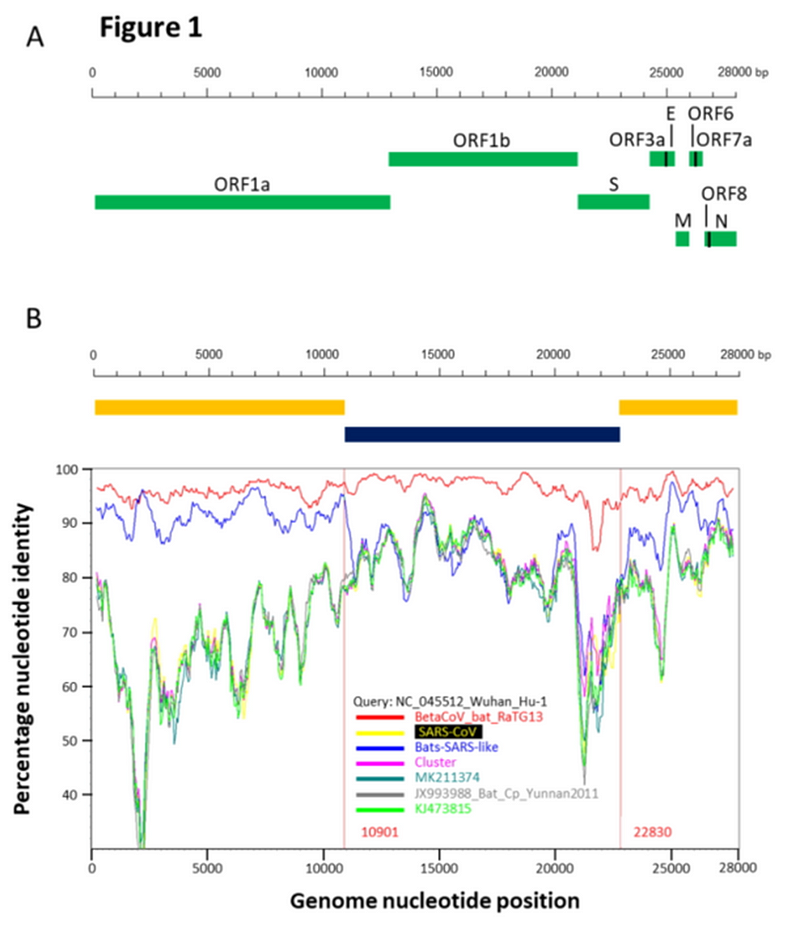
RaTG13 — это красная кривая, а синяя — это наиболее близкие к RaTG13 штаммы (ZXC21 и ZC45). Эти штаммы были выделены из Китайских подковоносов (Rhinolophus sinicus) в Чжоушане в 2015 (ZXC21) и 2017 (ZC45) годах. Как видно из графика, даже они очень сильно расходятся с RaTG13 (красная кривая) в районе S белка. При этом график выше не так хорошо передаёт масштаб пропасти между ними как прямое сравнение сиквенсов:
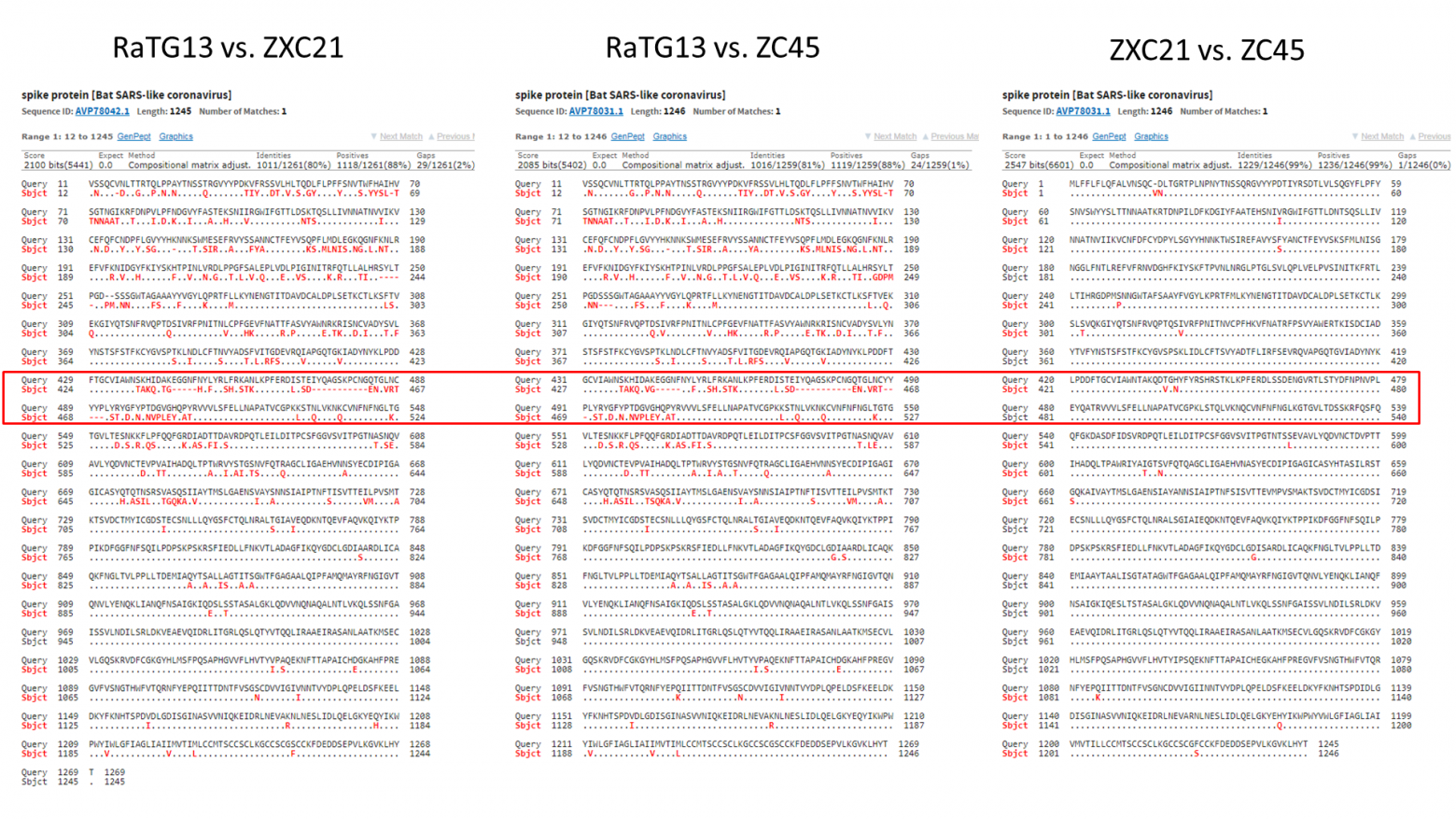
Как видим, шиповидные белки ZXC21 и ZC45 не только, в целом, на 23–24 аминокислотных остатка короче белка RaTG13, но они короче в самом важном месте — в RBM (см. делеции в красном прямоугольнике, отмеченные красными прочерками).
Так откуда же взялся RaTG13? Как я уже сказал, в 2020 году Ши Чжэнли сообщила, что выделила его у подковоносов (только вида Rhinolophus affinis, а не R. sinicus) в июле 2013 года в Юньнани. Правда, до конца января 2020 года публично о его существовании не сообщалось, а вот как описывает своё знаменательное открытие о его схожести с CoV2 сама группа Ши Чжэнли:
Затем мы обнаружили, что короткий участок РНК-зависимой РНК-полимеразы (RdRp) от коронавируса летучей мыши (BatCoV RaTG13), который ранее был обнаружен у Rhinolophus affinis из провинции Юньнань, показал высокую идентичность последовательности 2019-CoV2. Мы провели полноразмерное секвенирование на этом образце РНК (регистрационный номер GISAID EPI_ISL_402131). Анализ Simplot показал, что 2019-CoV2 очень похож на RaTG13 (Fig. 1c), с общей идентичностью генома 96,2%.Негусто: ну был у них этот штамм «ранее обнаружен», и был. Лежал на полке. До 2020 года секвенировали в нём только часть генома, отвечающую за RdRp. Откуда он на полке взялся? В Юньнани в 2013 году выделили. Где именно? Не сказали. В GenBank тоже не было этой информации. Но, к счастью, в базе геномов GISAID была: сообщается, что в городе Пуэр (да, на родине любимого чая Басты) его выделили из fecal swab некого самца:
Исходный текстWe then found that a short region of RNA-dependent RNA polymerase (RdRp) from a bat coronavirus (BatCoV RaTG13) — which was previously detected in Rhinolophus affinis from Yunnan province — showed high sequence identity to 2019-CoV2. We carried out full-length sequencing on this RNA sample (GISAID accession number EPI_ISL_402131). Simplot analysis showed that 2019-CoV2 was highly similar throughout the genome to RaTG13 (Fig. 1c), with an overall genome sequence identity of 96.2%.

Это меня немного заинтриговало, потому что в моих блужданиях по Пабмеду, я уже натыкался на экспедицию в Пуэр летом 2013 года:
Летучие мыши были пойманы в различных местах в пяти округах четырех префектур провинции Юньнань, Китай, с мая по июль 2013 года.
Исходный текстBats were captured from various locations in five counties of four prefectures of Yunnan Province, China, from May to July 2013.
В той экспедиции ничего особо интересного для нас исследователи не нашли, но, быть может, именно тогда Ши Чжэнли (или кто-то из её группы?) выделил тот самый образец RaTG13? Который они секвенировали лишь частично, но почему-то решили не публиковать, хотя он весьма отличался от всего известного ранее.
Сама Ши Чжэнли вполне могла лично участвовать в той экспедиции, потому что о таких экспедициях она отзывалась весьма тепло — например, в своём TED-like выступлении в 2018 году, где она показывала свои фотографии из таких экспедиций:
Именно серия таких экспедиций и принесла ей мировую славу и прозвище «Бэтвумен»: в 2013 году в статье в Nature группа Ши Чжэнли триумфально объявила о том, что в пещерах Юньнани она обнаружила летучих мышей-носителей штаммов RsSHC014 и Rs3367, которые совпадали с первым SARS-CoV на 85% и 96% соответственно.
Кстати, забавное совпадение, что примерно в то же время, в той же Юньнани группа Ши Чжэнли, оказывается, также обнаружила и штамм RaTG13, который оказался наиболее близок к CoV2, и их геномы тоже совпадают на 96%.
«Ухань-1»
Возвращаясь к той триумфальной статье от 2013 года, в ней же группа Ши Чжэнли ещё рассказала, что с помощью культивирования выделенных образцов в культуре обезьяньих клеток Vero, им удалось выделить живой вирус, который был практически идентичен штамму Rs3367 (наиболее близкому к SARS-CoV). Своё детище авторы окрестили WIV1 (где WIV обозначает Wuhan Institute of Virology):
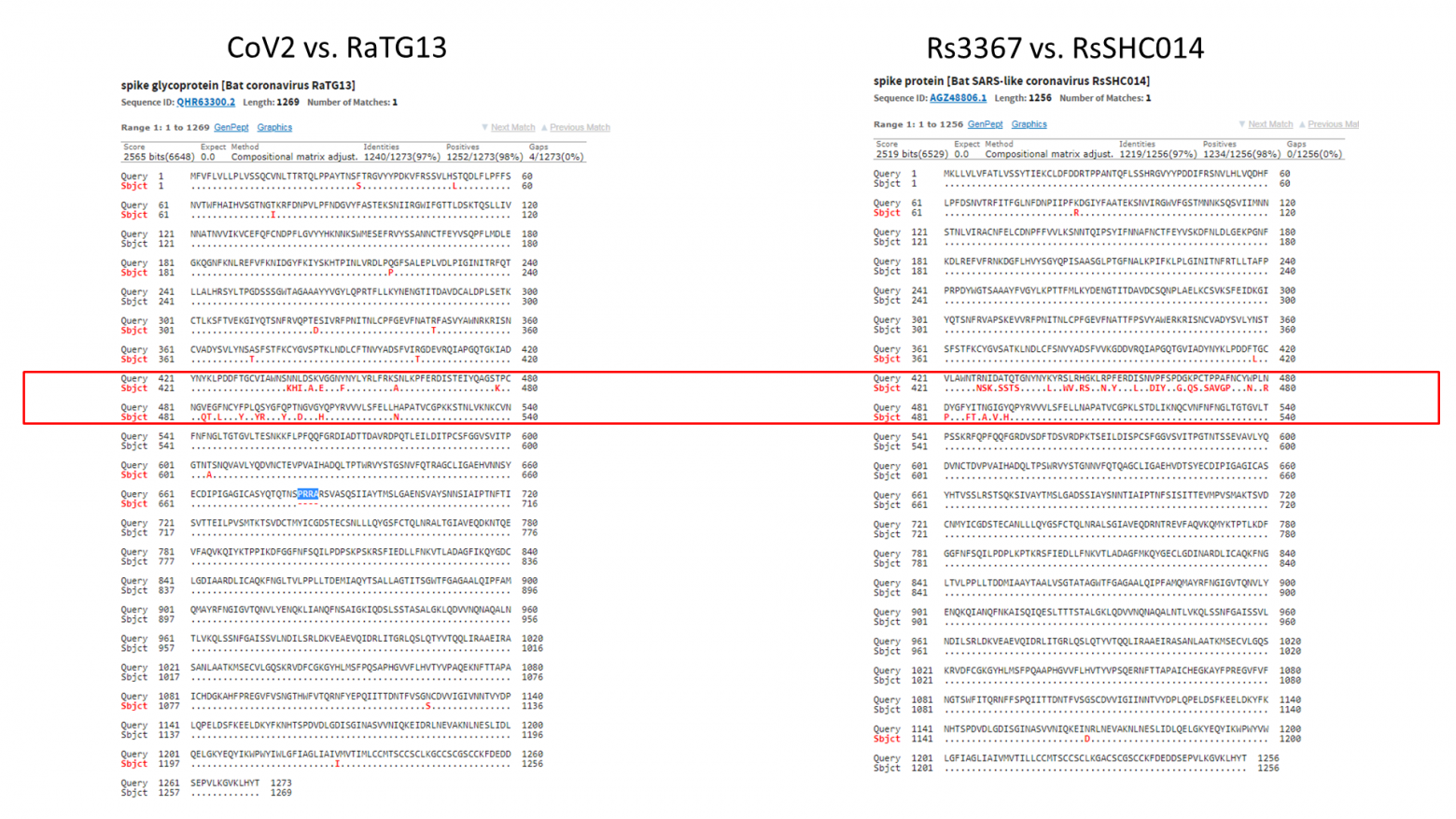
Что наиболее важно, мы сообщаем о первой изоляции живого вируса SL-CoV (летучемышиный SL-CoV-WIV1) из образцов, выделенных из фекалий летучей мыши, в клетках Vero E6, который имеет типичную морфологию коронавируса, идентичность генома на 99,9% с Rs3367 и использует ACE2 людей, цивет и подковоносов для проникновения в клетку. Предварительное тестирование in vitro показывает, что WIV1 также имеет широкий видовой тропизм.Давайте сравним RaTG13 со штаммами Rs3367 и RsSHC014:
Исходный текстMost importantly, we report the first recorded isolation of a live SL-CoV (bat SL-CoV-WIV1) from bat faecal samples in Vero E6 cells, which has typical coronavirus morphology, 99.9% sequence identity to Rs3367 and uses ACE2 from humans, civets and Chinese horseshoe bats for cell entry. Preliminary in vitro testing indicates that WIV1 also has a broad species tropism.

Как видим, шиповидный белок этих штаммов не только короче RaTG13-шного на 13 аминокислот, но и сильно отличается он него в первой его четверти. Кстати, любопытно, что шиповидные белки у Rs3367 (aka WIV1) и RsSCH014 практически идентичны, и различаются только в районе RBD (правый сиквенс ниже). Почти как CoV2 и RaTG13 (не считая фуриновой врезки):
Мог ли какой-нибудь исследователь, получив в марте 2019-го образцы коронавируса из конфискованных таможней панголинов, захотеть проверить, насколько панголиний RBM тропен к человеческому рецептору? А если ещё и с polybasic фуриновым сайтом в самом интересном месте? Вот это будет бомба!
Теоретически, конечно, мог бы. Ничего технически сложного для вирусологов в осуществлении таких экспериментов нет. Резонный вопрос: а зачем им для этого использовать RaTG13 в качестве основы, а не уже опробованный WIV1? Но вполне возможно, что химеру с WIV1 они тоже тестировали. А параллельно решили смоделировать вариант рекомбинации панголиньего вируса с более близким ему летучемышиным — ведь RaTG13 всё же ближе к панголиньим штаммам чем WIV1: его шиповидный белок ближе к ним и филогенетически, и структурно — он с ними даже по длине совпадает, в то время как белки WIV1/Rs3367 и RsSHC014 на 13 аминокислот короче панголиньих. Да и общая для RaTG13 и панголина-19 (MP789) мутация QTQTNS в районе протеазного сайта не может оставить равнодушной специалиста.
Другие юньнаньские штаммы
Кстати, другие исследователи в 2011 году тоже находили образцы коронавирусов у Юньнаньских Rhinolophus affinis.Самым интересным мне показался штамм LYRa11:
Но и он весьма далёк от RaTG13, а куда ближе к Rs3367 (это тот штамм, что на 96% совпадает с первым SARS-CoV):

А вот RaTG13, выделенный из тех же летучих мышей Rhinolophus affinis, что и LYRa11, на него похож меньше всего (левый бласт).
Ну и наконец, ещё один Юньнаньский штамм (бесхитростно названный Yunnan2011), выделенный в 2011 году из другого подвида подковоносов, Rhinolophus pusillus, похож на RaTG13 ещё меньше чем LYRa11:

Да и между собой Yunnan2011 и LYRa11 (правый бласт выше) не особо похожи, не считая высококонсервативную область S2. Кстати, что за бардак у них с названиями штаммов? То полностью год прописывают, то частично, а то и вовсе нет (Rs3367). То сначала идёт вид носителя (RaTG13), то потом (LYRa11). Ещё вот интересно, что обозначают TG, LY или SHC? Инициалы расшифровавшего геном?
Ладно, перейдём уже от вирусной археологии к вирусной инженерии, а именно к пересадке ключевых участков шиповидного белка и прочим gain-of-function (GOF) экспериментам.
1999: Первый химерный коронавирус
Eсли вы думаете, что вся эта GOF-движуха с анализом того, что именно позволяет коронавирусам перепрыгивать с одного вида на другой началась в ответ на эпидемию первого SARS-а в 2002 году, вы ошибаетесь. Вирусологи экспериментировали с химерными коронавирусами задолго до этого. Вот, например, показательная статья от 1999 года от голландской группы Питера Роттиера из Утрехта с говорящим названием “Ретаргетинг коронавируса путем замены эктодомена в шиповидном белке: пересечение видового барьера”:
Используя направленную РНК-рекомбинацию, мы сконструировали мутант вируса коронавирусного мышиного гепатита (MHV), в котором эктодомен шиповидного белка был заменен высоко дивергентным эктодоменом белка вируса инфекционного перитонита кошек. Полученный в результате химерный вирус, обозначенный fMHV, приобрел способность инфицировать клетки кошачьих и одновременно утратил способность заражать мышиные клетки в культуре.Кстати, похоже, что Ши Чжэнли стажировалось под руководством Питера Роттиера в Утрехте. По крайней мере, в 2005-ом она была в соавторах совместной статьи, где Утрехт был указан в качестве её аффилиации (но текущий адрес был уже указан в Шанхайском институте). Кстати, сама статья тоже весьма любопытна — в ней авторы исследовали что именно позволяет вирусам расширять свой видовой тропизм:
Исходный текстUsing targeted RNA recombination, we constructed a mutant of the coronavirus mouse hepatitis virus (MHV) in which the ectodomain of the spike glycoprotein (S) was replaced with the highly divergent ectodomain of the S protein of feline infectious peritonitis virus. The resulting chimeric virus, designated fMHV, acquired the ability to infect feline cells and simultaneously lost the ability to infect murine cells in tissue culture.
Лишь небольшое количество мутаций в его шиповидном белке позволяет мышиному коронавирусу переходить от мышино-ограниченного тропизма к расширенному диапазону хозяев путем пассирования in vitro. Один такой вирус, который мы изучали, приобрел два предполагаемых гепарансульфатсвязывающих сайта, сохранив при этом фуриновый сайт в другом месте.Во-первых, любопытно, что фуриновый сайт в том вирусе (SRRAHR|SV) похож на сайт в CoV2 (SPRRAR|SV), хотя у CoV2 он режется более эффективно из-за сдвоенных аргининов (это и делает его polybasic сайтом, то есть у него в последовательности RxxR несколько основных аминокислот подряд):
Исходный текстOnly a relatively few mutations in its spike protein allow the murine coronavirus to switch from a murine-restricted tropism to an extended host range by being passaged in vitro. One such virus that we studied had acquired two putative heparan sulfate-binding sites while preserving another site in the furin-cleavage motif.
Но особенно статья любопытна тем, что мутации, позволившие вирусу “расширить свой кругозор”, произошли даже не в лабораторных животных, а в пробирке (by being passaged in vitro). Причем, похоже, происходили они довольно быстро:
MHV/pi23 — вирус, полученный после 23 из 600 пассажей, приведших к MHV/BHK, также содержит предполагаемый HS-связывающий сайт в домене S1 в том же положении, что и в MHV/BHK, хотя и в виде меньшей вставки, в то время как у него отсутствует предполагаемый HS-связывающий сайт непосредственно перед пептидом слияния. MHV/pi23 способен инфицировать не мышиные клетки, хотя и менее эффективно, чем MHV/BHK. В дополнение ко множественным HS-связывающим сайтам, однако, мутации, обнаруженные в других частях белка S, таких как домен HR1 и предполагаемый пептид слияния (Fig. 1), также могут способствовать эффективному проникновению в немуринные клетки. В настоящее время мы находимся в процессе определения мутаций белка S, которые необходимы для фенотипа расширенного диапазона хозяев.Забегая вперёд, упомяну, что были и другие группы, которые пытались мутациями в пробирке увеличить вирулентность коронавируса, например MERS-а:
Исходный текстMHV/pi23, a virus obtained after 23 of the 600 passages that resulted in MHV/BHK, also contains a putative HS-binding site in the S1 domain at the same position as in MHV/BHK, albeit as a smaller insertion, while it lacks the putative HS-binding site immediately upstream of the fusion peptide. MHV/pi23 does infect nonmurine cells to some extent but much less efficiently than MHV/BHK. In addition to the multiple HS-binding sites, however, mutations found in other parts of the S protein, such as the HR1 domain and the putative fusion peptide (Fig. 1), might also contribute to the efficient entry into nonmurine cells. We are currently in the process of determining the S protein mutations that are required for the extended host range phenotype.
Чтобы лучше понять адаптивность видов MERS-CoV, мы использовали субоптимальный вариант DPP4 для изучения вирусной адаптации. Пассирование вируса на клетках, экспрессирующих этот вариант DPP4, приводило к накоплению мутаций в вирусном шиповидном белке, которые увеличивали репликацию.Причём у них мутации возникали уже через несколько пассажей (раундов размножения клеточных культур):
Исходный текстTo better understand the species adaptability of MERS-CoV, we identified a suboptimal species-derived variant of DPP4 to study viral adaption. Passaging virus on cells expressing this DPP4 variant led to accumulation of mutations in the viral spike which increased replication.
(F) Схема появления одиночных и двойных мутаций в шиповидном белке MERS-CoV в разных пассажах.Но это всё будет гораздо позже. А пока давайте вернёмся в 2002 год — ещё ДО вспышки первого SARS-CoV.
(G) Расположение мутаций в шиповидном белке MERS-CoV.
Исходный текст(F) Schematic of single and double mutation emergence in MERS-CoV spike over different passages.
(G) Location of mutations within MERS-CoV spike.
Как Барик всем путь озарил
Ральф Барик — это человек-легенда в коронавирусологии. Настоящий первопроходец синтетических методик манипуляции вирусных геномов. Ещё в 2002 году он опубликовал прорывную работу, открывшую новую веху как в изучении различных механизмов природных вирусов, так и в gain-of-function (GOF) исследованиях. В своей работе группа Барика синтетически воссоздала клон природного мышиного коронавируса:
Был разработан новый метод сборки полноразмерной инфекционной кДНК штамма вируса гепатита мыши коронавируса II группы А59 (MHV-A59). Были выделены семь сегментов кДНК, которые охватывали весь геном MHV размером 31,5 тысяч пар нуклеотидов. Концы кДНК были сконструированы с уникальными стыками и собраны только с соседними субклонами кДНК, в результате чего была получена интактная конструкция кДНК MHV-A59 длиной ~ 31,5 т.п.н. Сайты рестриктаз в местах стыков, которые располагались на концах каждого сегмента кДНК, систематически удалялись во время сборки полного полноразмерного продукта кДНК, что позволяет проводить повторную сборку без внесения нуклеотидных изменений… Этот метод потенциально может быть использован для конструирования вирусных, микробных или эукариотических геномов, достигающих нескольких миллионов пар оснований в длину и использоваться для вставки сайтов рестрикции в любом заданном месте в микробном геноме.
Исходный текстA novel method was developed to assemble a full-length infectious cDNA of the group II coronavirus mouse hepatitis virus strain A59 (MHV-A59). Seven contiguous cDNA clones that spanned the 31.5-kb MHV genome were isolated. The ends of the cDNAs were engineered with unique junctions and assembled with only the adjacent cDNA subclones, resulting in an intact MHV-A59 cDNA construct of ?31.5 kb in length. The interconnecting restriction site junctions that are located at the ends of each cDNA are systematically removed during the assembly of the complete full-length cDNA product, allowing reassembly without the introduction of nucleotide changes… The method has the potential to be used to construct viral, microbial, or eukaryotic genomes approaching several million base pairs in length and used to insert restriction sites at any given nucleotide in a microbial genome.
То есть авторы, по сути, перевели РНК-вирус на язык ДНК (с помощью обратной транскриптазы), для того чтобы затем им было удобно манипулировать его геномом с помощью имеющихся инструментов генной инженерии. Создав 7 таких cDNA сегментов провируса, авторы затем их сшили, причем «бесшовно» (не оставляя следов), после чего перевели свою конструкцию обратно в РНК, из которой затем в других клетках сформировались вирусные частицы.
SARS-2003
Буквально спустя несколько недель после публикации той работы группы Барика грянула эпидемия SARS-CoV, и Ральф Барик не терял времени даром. Уже летом 2003 его группа отправила в печать работу по созданию синтетического клона SARS-CoV:
Используя набор смежных сегментов кДНК, которые охватывают весь геном, мы собрали полноразмерную кДНК штамма SARS-CoV Urbani и выделили синтетические клоны вирусы SARS (инфекционный клон SARS-CoV), которые содержали ожидаемые маркерные мутации, вставленные в клоны. Рекомбинантные вирусы реплицировались так же эффективно, как вирус дикого типа, и оба были ингибированы обработкой ингибитором цистеинпротеиназы… Наличие полноразмерной кДНК SARS-CoV создаёт инструмент для манипулирования вирусным геномом, позволяя быстро и рационально разрабатывать и тестировать кандидаты вакцин и терапевтических средств против этого важного человеческого патогена.
Исходный текстUsing a panel of contiguous cDNAs that span the entire genome, we have assembled a full-length cDNA of the SARS-CoV Urbani strain, and have rescued molecularly cloned SARS viruses (infectious clone SARS-CoV) that contained the expected marker mutations inserted into the component clones. Recombinant viruses replicated as efficiently as WT virus and both were inhibited by treatment with the cysteine proteinase inhibitor… Availability of a SARS-CoV full-length cDNA provides a template for manipulation of the viral genome, allowing for the rapid and rational development and testing of candidate vaccines and therapeutics against this important human pathogen.
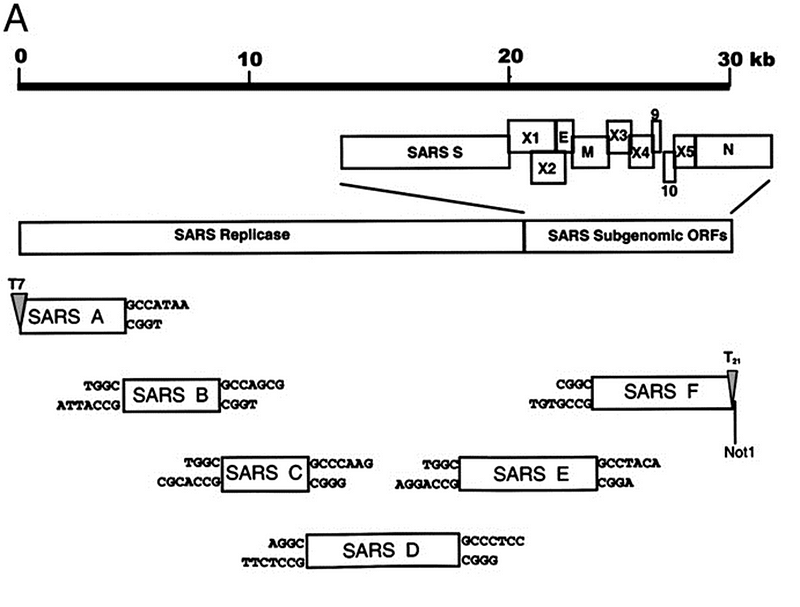
Скорость группы Барика позволяет понять, как быстро квалифицированная команда вирусологов может создать синтетический клон из природного вируса, а значит и вносить в него генетические модификации. Причём это было в 2003 году. Сегодня всё то же самое квалифицированная лаборатория может повторить за считанные недели.
SARS-2006
Барик был первым, но далеко не последним. Генная инженерия развивалась семимильными шагами, создавая всё новые и новые инструменты. Другие группы отрабатывали альтернативные технологии синтетической вирусологии. Например, в 2006 испанские исследователи повторили достижения Барика, тоже создав синтетический клон SARS, но с помощью другой технологии (искусственной бактериальной хромосомы):
В данном исследовании описана разработка полноразмерного инфекционного клона кДНК и функционального репликона штамма Urbani коронавируса SARS-CoV с помощью бактериальных искусственных хромосом (BAC). В этой системе вирусная РНК экспрессировалась в ядре клетки под контролем промотора цитомегаловируса и затем амплифицировалась в цитоплазме вирусной репликазой. И инфекционный клон, и репликон были полностью стабильны в Escherichia coli.
…
Собранный инфекционный клон кДНК SARS-CoV был полностью стабилен при размножении в клетках E.coli DH10B в течение более 200 поколений, что значительно облегчало генетические манипуляции с вирусным геномом (данные не показаны). Подробная стратегия клонирования и плазмидные последовательности доступны по запросу.
Исходный текстThe engineering of a full-length infectious cDNA clone and a functional replicon of the severe acute respiratory syndrome coronavirus (SARS-CoV) Urbani strain as bacterial artificial chromosomes (BACs) is described in this study. In this system, the viral RNA was expressed in the cell nucleus under the control of the cytomegalovirus promoter and further amplified in the cytoplasm by the viral replicase. Both the infectious clone and the replicon were fully stable in Escherichia coli.
…
The assembled SARS-CoV infectious cDNA clone was fully stable during its propagation in E. coli DH10B cells for more than 200 generations, considerably facilitating the genetic manipulation of the viral genome (data not shown). The detailed cloning strategy, plasmid maps, and sequences are available upon request.
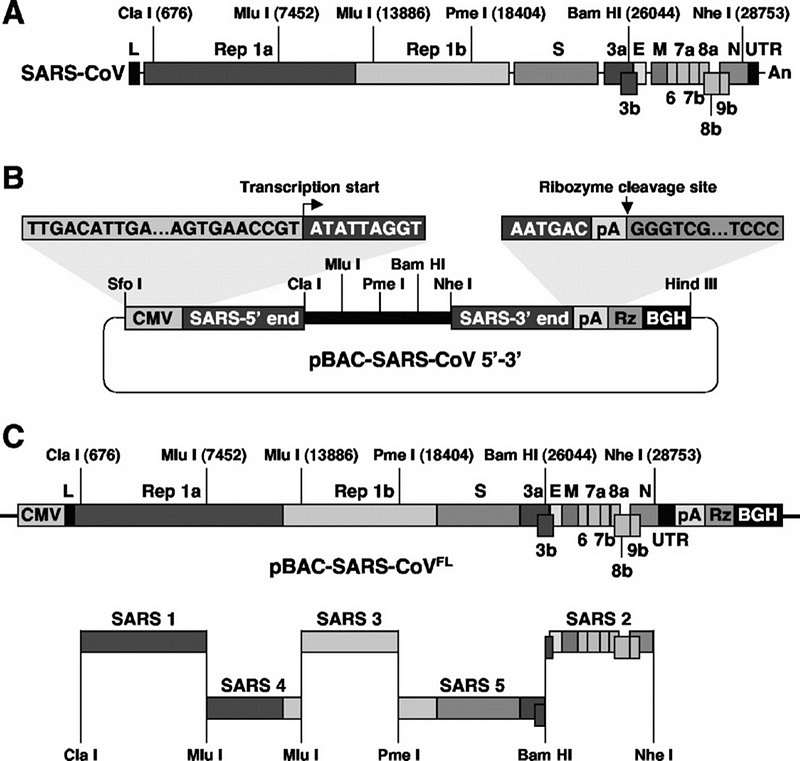
Правда, сделали они это не так элегантно как Барик, так как в финальной сборке синтетического вируса у них остались добавленные сайты рестриктаз, в то время как Барик научился соединять фрагменты «бесшовно». Но это мелочи, подход испанцев тоже вполне рабочий — в 2013 году с его помощью они создали синтетический клон MERS-a, а в 2015 году их методика вошла в учебник-справочник по коронавирусам (глава 13):

Ухань-2007
Но вернёмся в 2007 год. Тогда в гонку синтетической вирусологии включилась и группа Ши Чжэнли с работой, изучавшей шиповидный белок человеческого и летучемышиного коронавирусов, пытаясь определить, что именно в нём отвечает за способность перескакивать с вида на вид:
Ряд химерных шиповидных белков был сконструирован путем вставки различных последовательностей шиповидного белка SARS-CoV в основу из белка SL-CoV.То есть авторы вставляли разные отрезки из белка человеческого SARS-CoV в белок летучемышиного вируса. Вот их вывод:
Исходный текстA series of S chimeras was constructed by inserting different sequences of the SARS-CoV S into the SL-CoV S backbone.
Из этих результатов было установлено, что область от 310 до 518 в шиповидном белке BJ01 была необходимой и достаточной для получения шиповидным белком Rp3 способности связывания с человеческим ACE2.При этом они пробовали заменять и более короткие фрагменты, включая только RBM:
Исходный текстFrom these results, it was deduced that the region from aa 310 to 518 of BJ01-S was necessary and sufficient to convert Rp3-S into a huACE2-binding molecule.
Для вставки RBM из SARS-CoV в шиповидный белок SL-CoV, использовалась кодирующая область от 424 до 494 аминокислоты в шиповидном белке BJ01 для замены соответствующих областей в белке Rp3, в результате чего был получен химерный ген шиповидного белка (CS), обозначенный как CS424–494.С учётом того, что это было написано в 2007 году, думаю, сегодня произвести замену RBM одного вируса на другой не составит труда даже начинающему вирусологу.
Исходный текстFor introduction of the RBM of SARS-CoV S into the SL-CoV S, the coding region from aa 424 to 494 of BJ01-S was used to replace the corresponding regions of Rp3-S, resulting in a chimeric S (CS) gene designated CS424–494.
Химера-2015
В свете вышеописанных экспериментов, не очень понятно, чем именно был вызван тот фурор, которая произвела, наверное, самая нашумевшая gain-of-function публикация. Речь, конечно же, о совместной работе Ши Чжэнли и Ральфа Барика, в которой они создали синтетический химерный вирус:
Используя систему обратной генетики SARS-CoV, мы создали и охарактеризовали химерный вирус, экспрессирующий шиповидный белок коронавируса летучей мыши SHC014 в мышиноадаптированной основе SARS-CoV. Результаты показывают, что вирусы группы 2b, кодирующие шиповидный белок SHC014 в основе дикого типа, могут эффективно использовать несколько ортологов человеческого рецептора SARS (ACE2), эффективно реплицироваться в первичных клетках дыхательных путей человека и достигать титров in vitro, эквивалентных эпидемическим штаммам SARS-CoV. Кроме того, эксперименты in vivo демонстрируют репликацию химерного вируса в легких мыши с заметным патогенезом. Оценка доступных иммунотерапевтических и профилактических методов на основе атипичной пневмонии показала низкую эффективность; подходы как к моноклональным антителам, так и к вакцинам не смогли нейтрализовать и защитить от инфицирования CoV с новым шиповидным белком. На основании этих результатов мы синтетически воспроизвели инфекционный полноразмерный рекомбинантный вирус SHC014 и продемонстрировали репликацию вируса как in vitro, так и in vivo.То есть, по сути, исследователи шли уже проторённой дорогой: взяли шиповидный белок из RsSHC014, который Ши Чжэнли выделила из юньнаньских летучих мышей в 2011 году, и вставили его в SARS-CoV, специально адаптированный под рождённых ползать мышей, для последующих in vivo экспериментов на этих самых мышах. Ну и на человеческих клетках новую конструкцию проверили. А заодно поупражнялись в создании рекомбинантного клона того же самого RsSHC014 — ну а почему бы и нет? Ведь, во-первых, это красиво:
Исходный текстUsing the SARS-CoV reverse genetics system, we generated and characterized a chimeric virus expressing the spike of bat coronavirus SHC014 in a mouse-adapted SARS-CoV backbone. The results indicate that group 2b viruses encoding the SHC014 spike in a wild-type backbone can efficiently use multiple orthologs of the SARS receptor human angiotensin converting enzyme II (ACE2), replicate efficiently in primary human airway cells and achieve in vitro titers equivalent to epidemic strains of SARS-CoV. Additionally, in vivo experiments demonstrate replication of the chimeric virus in mouse lung with notable pathogenesis. Evaluation of available SARS-based immune-therapeutic and prophylactic modalities revealed poor efficacy; both monoclonal antibody and vaccine approaches failed to neutralize and protect from infection with CoVs using the novel spike protein. On the basis of these findings, we synthetically re-derived an infectious full-length SHC014 recombinant virus and demonstrate robust viral replication both in vitro and in vivo.
(a) Схема молекулярного клона SHC014-CoV, который был синтезирован в виде шести смежных сегментов кДНК (обозначенных как SHC014A, SHC014B, SHC014C, SHC014D, SHC014E и SHC014F), фланкированных уникальными рестриктазными сайтами BglI, которые обеспечили направленную сборку полноразмерной экспрессии кДНК (для 1a, 1b, спайка, 3, конверт, матрица, 6–8 и нуклеокапсид). Подчеркнутые нуклеотиды представляют собой выступающие последовательности, образованные после расщепления рестриктазой.А во-вторых, исследователи поняли, что не только тропностью шиповидного белка к рецептору определяется потенциал вируса к переходу из одного вида животных на другой — потому что химера SHC014-MA15 была более вирулентной чем сам SHC014, даже в человеческих клетках:
Исходный текст(a) Schematic of the SHC014-CoV molecular clone, which was synthesized as six contiguous cDNAs (designated SHC014A, SHC014B, SHC014C, SHC014D, SHC014E and SHC014F) flanked by unique BglI sites that allowed for directed assembly of the full-length cDNA expressing open reading frames (for 1a, 1b, spike, 3, envelope, matrix, 6–8 and nucleocapsid). Underlined nucleotides represent the overhang sequences formed after restriction enzyme cleavage.
Примечательно, что дифференциальный тропизм в легких по сравнению с таковым у SARS-MA15 и ослабление полноразмерного SHC014-CoV в культурах [эпителиальных дыхательных путей человека] относительно SARS-CoV Urbani позволяют предположить, что помимо связывания с ACE2 и другие факторы — включая процессивность шиповидного белка, биодоступность рецептора или антагонизм иммунных ответов хозяина — могут способствовать вирулентности.Особо хочу выделить процессивность шиповидного белка в цитате, потому что это далеко не первый раз когда исследователи писали, что способность шиповидного белка к расщеплению протеазами (включая фурин) оказывает значительное влияние на вирулентность.
Исходный текстNotably, differential tropism in the lung as compared to that with SARS-MA15 and attenuation of full-length SHC014-CoV in [human epithelial airway cell] cultures relative to SARS-CoV Urbani suggest that factors beyond ACE2 binding — including spike processivity, receptor bio-availability or antagonism of the host immune responses — may contribute to emergence.
В заключение темы — совместное фото Ральфа Барика и Ши Чжэнли. Фото сделано в Ухане, в октябре 2018-го:

Мышиный SARS-2007
Тут не могу не упомянуть, что это был за “мышиный вирус MA15” в предыдущей работе. Это вовсе не какой-то природный мышиный коронавирус, как можно было бы предположить. А это лабораторно модифицированный человеческий SARS-CoV, который ещё в 2007-ом та же группа Барика — видимо соревнуясь с группой Ши Чжэнли (помните их статью от 2007 года) – превратила в настоящего мышиного убийцу. Для этого они его сначала итеративно “улучшали” на мышах, а когда он через несколько итераций стал максимально “эффективным”, они воспроизвели возникшие в мышах мутации в синтетическом клоне нового вируса, и ещё раз проверили, что он действительно обладает повышенной инфективностью:
Мы адаптировали SARS-CoV (штамм Urbani) путем серийного пассирования в дыхательных путях молодых мышей BALB/c. Пятнадцать пассажей привели к вирусу (MA15), который является летальным для мышей после интраназальной инокуляции. Смертельности предшествует быстрая репликация вируса с высоким титром в легких, виремия и распространение вируса на внелёгочные участки, сопровождающиеся лимфопенией, нейтрофилией и патологическими изменениями в легких. Обильный вирусный антиген широко распространен в эпителиальных клетках бронхов и альвеолярных пневмоцитах, а некротический клеточный дебрис присутствует в дыхательных путях и альвеолах с легким и очаговым пневмонитом. Эти наблюдения показывают, что мыши, инфицированные МА15, умирают от подавляющей вирусной инфекции с обширным, опосредованным вирусом разрушением пневмоцитов и реснитчатых эпителиальных клеток. Вирус MA15 имеет шесть кодирующих мутаций, связанных с адаптацией и повышенной вирулентностью; при введении в рекомбинантный SARS-CoV эти мутации приводят к высоковирулентному и летальному вирусу (rMA15), дублирующему фенотип биологически полученного вируса MA15. Интраназальная инокуляция МА15 воспроизводит многие аспекты заболевания, наблюдаемые в тяжелых случаях SARS у человека.
Исходный текстWe adapted the SARS-CoV (Urbani strain) by serial passage in the respiratory tract of young BALB/c mice. Fifteen passages resulted in a virus (MA15) that is lethal for mice following intranasal inoculation. Lethality is preceded by rapid and high titer viral replication in lungs, viremia, and dissemination of virus to extrapulmonary sites accompanied by lymphopenia, neutrophilia, and pathological changes in the lungs. Abundant viral antigen is extensively distributed in bronchial epithelial cells and alveolar pneumocytes, and necrotic cellular debris is present in airways and alveoli, with only mild and focal pneumonitis. These observations suggest that mice infected with MA15 die from an overwhelming viral infection with extensive, virally mediated destruction of pneumocytes and ciliated epithelial cells. The MA15 virus has six coding mutations associated with adaptation and increased virulence; when introduced into a recombinant SARS-CoV, these mutations result in a highly virulent and lethal virus (rMA15), duplicating the phenotype of the biologically derived MA15 virus. Intranasal inoculation with MA15 reproduces many aspects of disease seen in severe human cases of SARS.
Барик-2008
Кстати, о соперничестве Барика и Ши Чжэнли. Параллельно с пересадкой RBD от человеческого SARS-CoV в мышиный, его группа создавала такие же химеры и с летучемышиными штаммами. В 2008 году группа Барика взяла летучемышиный штамм Bat-SCoV и заменила в нём RBD в шиповидном белке на RBD из человеческого SARS-а. То есть, по сути, повторила работу Ши Чжэнли от 2007 года, только не ограничившись псевдо-вирусами, а создав самый настоящий химерный коронавирус — причём не какой-то мышиный как в работе 2015 года, а самый что ни на есть человеческий. Своим достижением авторы явно гордились:
Здесь мы сообщаем о разработке, синтезе и выделению самой крупной синтетической реплицирующейся жизненной формы размером 29,7 килобаз — коронавируса летучей мыши (Bat-SCoV), подобного тяжелому острому респираторному синдрому (SARS), вероятного предшественника эпидемии SARS-CoV.
…
Чтобы проверить, являются ли RBDs Bat-SCoV и SARS-CoV взаимозаменяемыми, мы заменили RBD Bat-SCoV (аминокислоты 323–505) на RBD SARS-CoV (аминокислоты 319–518) (27, 28) (GenBank регистрационный номер FJ211860), имитирующий теоретическую рекомбинацию, которая может произойти во время смешанной инфекции in vivo (Fig. 1B).
Исходный текстHere, we report the design, synthesis, and recovery of the largest synthetic replicating life form, a 29.7-kb bat severe acute respiratory syndrome (SARS)-like coronavirus (Bat-SCoV), a likely progenitor to the SARS-CoV epidemic.
…
To test whether the RBDs of Bat-SCoV and SARS-CoV were interchangeable, we replaced the Bat-SCoV RBD (amino acid 323–505) with the SARS-CoV RBD (amino acid 319–518) (27, 28) (GenBank accession no. FJ211860), simulating a theoretical recombination event that might occur during mixed infection in vivo (Fig. 1B).
(B) Схематическое изображение, показывающее организацию шиповидных белков SARS-CoV и Bat-SCoV. Спроектированные белки изображены ниже с названием вируса слева. Bat-SRBD включает всю последовательность шиповидного белка Bat-SCoV, за исключением того, что RBD Bat-SCoV (аминокислоты Bat-SCoV 323–505) заменен на RBD SARS-CoV (аминокислоты 319–518) (инвентарный номер GenBank FJ211860 ). Bat-SRBD-MA включает изменение RBD в шиповидном белке MA15 на SARS-CoV Y436H. Bat-SRBM включает минимальные 13 остатков SARS-CoV, критических для контакта с ACE2, что приводит к химерному RBD с аминокислотами 323I-429T Bat-SCoV и аминокислотами 426R-518D SARS-CoV. Bat-Hinge представляет собой последовательность Bat-SRBM, где аминокислоты Bat-SCoV 392L-397E заменены на аминокислоты SARS-CoV 388V-393D. Bat-F включает нуклеотидную последовательность 1–24057 SARS-CoV (до аминокислоты 855 в шиповидном белке) с оставшейся 3'-последовательностью из Bat-SCoV. Справа от схематических изображений указаны наблюдаемые данные по транскрипционной активности и приблизительные титры на 1-м пассаже (P1). ND указывает, что инфекционный вирус не обнаружен.
Исходный текст(B) Schematic representation showing organization of the SARS-CoV and Bat-SCoV Spike proteins. The engineered Spike proteins are pictured below with the virus name to the left. Bat-SRBD includes all of the Bat-SCoV Spike sequence except that the Bat-SCoV RBD (Bat-SCoV amino acid 323–505) is replaced with the SARS-CoV RBD (amino acid 319–518) (GenBank accession no. FJ211860). Bat-SRBD-MA includes the MA15 Spike RBD change at SARS-CoV aa Y436H. Bat-SRBM includes the minimal 13 SARS-CoV residues critical for ACE2 contact, resulting in a chimeric RBD of Bat-SCoV amino acid 323I-429T and SARS-CoV amino acid 426R-518D. Bat-Hinge is Bat-SRBM sequence, with Bat-SCoV amino acid 392L-397E replaced with SARS-CoV amino acid 388V-393D. Bat-F includes nt 1–24057 of SARS-CoV (to Spike amino acid 855), with the remaining 3? sequence from Bat-SCoV. To the right of the schematic representations, observation of transcript activity and approximate stock titers at passage 1 (P1) are indicated. ND indicates no infectious virus detected by plaque assay.
Барик-2016
В творчестве Барика вообще много римейков. Например, в 2016-ом году он практически повторил ту самую совместную работу с Ши Чжэнли от 2015 года по созданию химерного вируса, только на этот раз он вставил в мышиноадаптированный SARS кусок шиповидного белка не из RsSCH014, а из другого найденного Ши Чжэнли в Юньнани его близкого родственника — Rs3367. Ну или, если быть совсем точным, то из штамма WIV1 — лабораторного клона Rs3367, выращенного в клеточной культуре в Уханьском институте вирусологии в 2013 году. Вот что именно сделала в 2016 году группа Барика:
Используя инфекционный клон SARS-CoV в качестве матрицы (7), мы разработали и синтезировали полноразмерный инфекционный клон WIV1-CoV, состоящий из шести плазмид, которые можно было бы ферментативно разрезать, лигировать вместе и электропорировать в клетки, для получения репликационно компетентных вирионов (рис. S1A). В дополнение к полноразмерному клону мы также создали химерный вирус WIV1-CoV, который заменил шиповидный белок в SARS на белок из WIV1 в адаптированной для мыши основе (WIV1-MA15, рис. S1B). … Для подтверждения кинетики роста и репликации клетки Vero были инфицированы SARS-CoV Urbani, WIV1-MA15 и WIV1-CoV.
Исходный текстUsing the SARS-CoV infectious clone as a template (7), we designed and synthesized a full-length infectious clone of WIV1-CoV consisting of six plasmids that could be enzymatically cut, ligated together, and electroporated into cells to rescue replication competent progeny virions (Fig. S1A). In addition to the full-length clone, we also produced WIV1-CoV chimeric virus that replaced the SARS spike with the WIV1 spike within the mouse-adapted backbone (WIV1-MA15, Fig. S1B). … To confirm growth kinetics and replication, Vero cells were infected with SARS-CoV Urbani, WIV1-MA15, and WIV1-CoV.
То есть, по большому счёту, в 2016 году Барик клонировал свою статью от 2015 года. Более того, её смысл мне не очень понятен: ведь WIV1/Rs3367, если помните, и так на 96% совпадал с SARS-CoV (за это Ши Чжэнли и получила известность). Поэтому зачем обратно в SARS-CoV вставлять шиповидный белок из его ближайшего родственника мне не очень ясно. Быть может, просто из любви к искусству. В этом свете заголовок его статьи приобретает некую двойственность: “SARS-подобный WIV1-CoV готов к переходу на человека”. Почему-то в голову сразу приходит этот кадр:

Ещё мне не понятно как в 2015 году Барик сумел получить патент на создание «химерных коронавирусных шиповидных белков», с учётом того, что всё это и он, и Ши Чжэнли публиковали задолго до 2015-го.
Барик-1990
Ладно, последний штрих к портрету Ральфа Барика. Он не просто старожил этой области, а занимался дизайном рекомбинантных коронавирусов ещё до всяких сиквенаторов и прочих современных инструментов генной инженерии. Вот его статья по созданию “температурных мутантов” из мышиного коронавируса от аж 1990-го года:
На протяжении всего этого исследования использовали штамм вируса гепатита мыши A59 (MHV-A59). Вирус размножали и клонировали три раза в непрерывной клеточной линии астроцитомы мыши (DBT).Так что созданием различных вирусных мутантов Барик занимается уже более 30 лет.
…
Различные комбинации температурно-чувствительных мутантов смешивали и инокулировали в клетки при множественности заражения, равной 10.
Исходный текстThe A59 strain of mouse hepatitis virus (MHV-A59) was used throughout the course of this study. Virus was propagated and cloned three times in the continuous murine astrocytoma cell line (DBT).
…
Various combinations of ts mutants were mixed and inoculated onto cells at a multiplicity of infection of 10 each.
Барик-2019
И, к слову сказать, даже сейчас темпов не снижает. В конце октября 2019-го его группа подала на публикацию очередную статью о важной роли фуринового сайта в шиповидном белке для преодоления коронавирусами «барьера для зоонозной инфекции»:
Вместе эти результаты демонстрируют, что протеазное расщепление также является основным барьером для инфекции клеток Vero с помощью HKU5-CoV. Рассматривая далее, мы сравнили предсказанное расщепление на границе S1/S2, S2’ и сайте эндосомной цистеиновой протеазы в шиповидных белках MERS, PDF2180 и HKU5 (Fig. 6D) (26). В сайте S1/S2 MERS, Uganda и HKU5 поддерживают сайт расщепления RXXR, хотя различные внутренние аминокислоты могут влиять на его эффективность. Для последовательности S2' MERS и HKU5 также сохраняют мотив RXXR; однако в шипах штамма Uganda отсутствует первый аргинин (SNAR), что потенциально влияет на его расщепление.Памятуя о соревновательном духе между группами Барика и Ши Чжэнли, интересно, какова вероятность, что в Ухане в конце 2019 года кто-то тоже занимался подобными исследованиями?
Исходный текстTogether, these results demonstrate that protease cleavage is also the primary barrier to infection of Vero cells with HKU5-CoV. Examining further, we compared the predicted cleavage at S1/S2 border, S2’, and the endosomal cysteine protease site across MERS, PDF2180, and HKU5 spikes (Fig. 6D) (26). For the S1/S2 site, MERS, Uganda, and HKU5 maintain the RXXR cleavage motif, although the different interior amino acids may alter efficiency. For the S2’ sequence, MERS and HKU5 also retain the RXXR motif; however, the Uganda spike lacks the first arginine (SNAR), potentially impacting cleavage.
Gain-of-Function: «Запретить нельзя продолжить»
Многие, кто впервые слышит о вышеописанных исследованиях, задаются резонным вопросом: «А нафига?» Зачем учёные создают химерные вирусы-убийцы? Политкорректный ответ: для разработки превентивной защиты (вакцин и препаратов) от возможных природных химер и для понимания рисков их возникновения. Вот, собственно, что сами Барик и Ши Чжэнли с соавторами писали на эту тему в той самой статье 2015 года:
В дополнение к подготовке против будущих появляющихся вирусов, этот подход должен быть рассмотрен в контексте предписанной правительством США паузы в исследованиях усиления функции (GOF). На основании предыдущих моделей (Fig. 4a, b) создание химерных вирусов, таких как SHC014-MA15, не ожидалось, что увеличится его патогенность. Хотя SHC014-MA15 аттенуирован по сравнению с его родительским мышиноадаптированным SARS-CoV, аналогичные исследования, изучавщие патогенность CoV с шипом Urbani дикого типа в основе MA15, не показали потери веса у мышей, показали сниженную репликацию вируса. Таким образом, по отношению к шиповидному белку Urbani-MA15 CoV, SHC014-MA15 демонстрирует усиление патогенеза (рис. 1). На основании этих результатов надзорные научные комитеты могут посчитать подобные исследования с созданием химерных вирусов, основанных на циркулирующих штаммах, слишком рискованными, поскольку нельзя исключать повышение патогенности в млекопитающих. В сочетании с ограничениями на адаптированные к мышам штаммы и разработкой моноклональных антител с использованием побочных мутантов, исследования появления новых коронавирусов и терапевтической эффективности могут быть серьезно ограничены в будущем. Вместе эти данные и ограничения представляют собой перекресток озабоченности GOF; потенциал для подготовки к будущим вспышкам и смягчения их последствий должен быть сопоставлен с риском создания более опасных патогенных микроорганизмов. При разработке политики в будущем важно учитывать ценность данных, полученных в результате этих исследований, и то, заслуживают ли эти типы исследований химерных вирусов продолжения в сравнении с присущими им рисками.Были ли эти слова пророческими? В конце 2014-го года США ввели мораторий на государственное финансирование таких gain-of-function исследований, но почти сразу (в 2017-ом) отменили. А в Китае никакого моратория на такие исследования не вводили, а даже наоборот открывали новые «супер(без)опасные лаборатории» уровня BSL-4, как в 2017 году в Ухане:
Исходный текстIn addition to offering preparation against future emerging viruses, this approach must be considered in the context of the US government–mandated pause on gain-of-function (GOF) studies. On the basis of previous models of emergence (Fig. 4a,b), the creation of chimeric viruses such as SHC014-MA15 was not expected to increase pathogenicity. Although SHC014-MA15 is attenuated relative to its parental mouse-adapted SARS-CoV, similar studies examining the pathogenicity of CoVs with the wild-type Urbani spike within the MA15 backbone showed no weight loss in mice and reduced viral replication. Thus, relative to the Urbani spike–MA15 CoV, SHC014-MA15 shows a gain in pathogenesis (Fig. 1). On the basis of these findings, scientific review panels may deem similar studies building chimeric viruses based on circulating strains too risky to pursue, as increased pathogenicity in mammalian models cannot be excluded. Coupled with restrictions on mouse-adapted strains and the development of monoclonal antibodies using escape mutants, research into CoV emergence and therapeutic efficacy may be severely limited moving forward. Together, these data and restrictions represent a crossroads of GOF research concerns; the potential to prepare for and mitigate future outbreaks must be weighed against the risk of creating more dangerous pathogens. In developing policies moving forward, it is important to consider the value of the data generated by these studies and whether these types of chimeric virus studies warrant further investigation versus the inherent risks involved.

На всякий случай, поясню, что до 2017 года лаборатория в Уханьском институте вирусологии была класса BSL-3, которого было достаточно для работы с коронавирусами. Приведу пару цитат из вышеприведённой заметки о создании уханьской BSL-4 лаборатории:
В планах на будущее — изучение патогена, вызывающего атипичную пневмонию, для которого также не требуется лаборатория BSL-4, прежде чем перейти к изучению вируса Эбола и западноафриканского вируса Ласса. Около миллиона китайцев работают в Африке; страна должна быть готова к любой ситуации, говорит Юань. «Вирусы не знают границ».Интересно, что помимо Уханя, новую BSL-4 лабораторию планировали открыть в Куньмине, причём с прицелом на тестирование вакцин на приматах. Куньмин, напомню, это столица Юньнани. Именно в близлежащих пещерах Ши Чжэнли обнаружила те самые штаммы Rs3367 и RsSHC014. Кстати, тестирование на приматах упоминалось в качестве возможных дальнейших шагов для разработки превентивных вакцин против потенциальных будущих вспышек коронавируса в «той самой» (сколько раз я её уже так назвал?) статье Барика и Ши Чжэнли:
…
План по расширению сети усиливает такие опасения. Одна лаборатория BSL-4 в Харбине уже ожидает аккредитации; следующие две, как ожидают, будут в Пекине и Куньмине, последняя сосредоточился на использовании обезьян для изучения болезней.
Лина говорит, что размер Китая оправдывает такой размах, и что возможность объединить исследования BSL-4 с обилием исследовательских обезьян — китайские исследователи сталкиваются с гораздо меньшим количеством бюрократических проволочек, чем на Западе, когда дело доходит до исследований на приматах — может быть мощной. «Если вы хотите протестировать вакцины или противовирусные препараты, вам нужна модель приматов, отличных от человека», — говорит Лина.
Но Эбрайт не убежден в необходимости более одной лаборатории BSL-4 в материковом Китае. Он подозревает, что расширение там является реакцией на сети лабораторий в Соединенных Штатах и Европе, что, по его словам, также неоправданно. Он добавляет, что правительства будут предполагать, что такая избыточная мощность предназначена для потенциального развития биологического оружия.
«Эти объекты по своей сути предназначены для двойного использования», — говорит он. Перспектива расширения возможностей для введения обезьянам патогенных микроорганизмов также беспокоит, а не вдохновляет его: «Они могут бегать, они могут царапать, они могут кусать».
Треван говорит, что инвестиции Китая в лабораторию BSL-4 могут, прежде всего, стать способом доказать миру, что страна конкурентоспособна. «Это большой статусный символ в биологии, — говорит он, — вне зависимости от того, нужны они или нет».
Исходный текстFuture plans include studying the pathogen that causes SARS, which also doesn’t require a BSL-4 lab, before moving on to Ebola and the West African Lassa virus, which do. Some one million Chinese people work in Africa; the country needs to be ready for any eventuality, says Yuan. “Viruses don’t know borders.”
…
The plan to expand into a network heightens such concerns. One BSL-4 lab in Harbin is already awaiting accreditation; the next two are expected to be in Beijing and Kunming, the latter focused on using monkey models to study disease.
Lina says that China’s size justifies this scale, and that the opportunity to combine BSL-4 research with an abundance of research monkeys — Chinese researchers face less red tape than those in the West when it comes to research on primates — could be powerful. “If you want to test vaccines or antivirals, you need a non-human primate model,” says Lina.
But Ebright is not convinced of the need for more than one BSL-4 lab in mainland China. He suspects that the expansion there is a reaction to the networks in the United States and Europe, which he says are also unwarranted. He adds that governments will assume that such excess capacity is for the potential development of bioweapons.
“These facilities are inherently dual use,” he says. The prospect of ramping up opportunities to inject monkeys with pathogens also worries, rather than excites, him: “They can run, they can scratch, they can bite.”
Trevan says China’s investment in a BSL-4 lab may, above all, be a way to prove to the world that the nation is competitive. “It is a big status symbol in biology,” he says, “whether it’s a need or not.”
Тем не менее, требуется дальнейшее тестирование на нечеловеческих приматах, чтобы перевести эти находки в патогенный потенциал у людей. Важно отметить, что недостаток доступных терапевтических средств определяет критическую необходимость дальнейшего изучения и разработки методов лечения. Обладая этими знаниями, можно разработать программы эпиднадзора, диагностические реагенты и эффективные методы лечения, которые могут защитить от появления коронавирусов, специфичных для группы 2b, таких как SHC014, и могут применяться к другим коронавирусным ветвям, которые поддерживают аналогичные гетерогенные пулы.Вполне возможно, что к 2019 году создание и тестирование потенциальных вакцин от различных SARS-like коронавирусов уже шло полным ходом.
Исходный текстHowever, further testing in nonhuman primates is required to translate these finding into pathogenic potential in humans. Importantly, the failure of available therapeutics defines a critical need for further study and for the development of treatments. With this knowledge, surveillance programs, diagnostic reagents and effective treatments can be produced that are protective against the emergence of group 2b–specific CoVs, such as SHC014, and these can be applied to other CoV branches that maintain similarly heterogeneous pools.
О сколько эпидемий чудных готовит просвещенья дух…
Ну что ж, давайте уже рассмотрим версию о лабораторной утечке. Для начала приведу краткую историческую справку о других утечках. Потому что побеги вирусов из лабораторий в прошлом случались неоднократно. В первую очередь, того же SARS-CoV: впервые он сбежал летом 2003 года в Сингапуре, потом в декабре 2003 на Тайване, а весной 2004 дважды утекал в Пекине.
Были тревожные звоночки и в Европе и США, хотя там обошлось без заражений. Например, во Франции лаборатория как-то потеряла пробирки с SARS-ом, а в США BSL-4 лаборатория в Техасе недосчитались пробирки с вирусом венесуэльской геморрагической лихорадки:
Только один ученый работал с вирусом, и Рейес сказал, что лаборатория подозревает, что ученый случайно выбросил пробирку в ноябре.История знает и другие, куда более широкомасштабные утечки. Например, «воскрешение» вируса гриппа H1N1 в 1977 году, который до этого считался исчезнувшим. Да, это вирус той самой «испанки»:
…
В Галвестонской биолаборатории самые строгие меры безопасности, потому что она изучает материалы уровня биологической безопасности BSL-4, то есть опасные инфекционные заболевания, которые не имеют вакцин или лекарств. Материалы BSL-4 включают Guanarito, Эболу и оспу.
Исходный текстOnly one scientist worked with the virus, and Reyes said the lab suspects that scientist accidentally threw the vial away in November.
…
Galveston biolab requires the most stringent safety measures because it studies biosafetly level BSL-4 materials, or dangerous infectious diseases that have no vaccines or cures. BSL-4 materials include Guanarito, Ebola and smallpox.
Вирусы человеческого гриппа H1N1 появились с пандемией 1918 года и сохранялись, накапливая небольшие изменения в геноме (с серьезными изменениями в 1947 году), пока в 1957 году не появился «азиатский» грипп H2N2, вызвавший всемирную пандемию. Вирус гриппа H1N1, по-видимому, вымер, и не выделялся в течение 20 лет. В 1969 году вирус H3N2 в Гонконге заменил вирус H2N2 и продолжает циркулировать.Похоже, создание температурно-чувствительных вирусных мутантов для разработки потенциальных «ослабленных» вакцин было широко распространено в конце ХХ века. Если помните, в 1990-м Барик тоже экспериментировал с созданием температурно-чувствительных штаммов.
В сентябре 1977 г. вирус гриппа H1N1 был выделен из больных в дальневосточном регионе Советского Союза, а в начале 1978 г. китайцы сообщили, что в мае 1977 г. они выделили вирус H1N1 на северо-востоке Китая, в регионе, прилегающем к месту советской вспышки. Используя ранние генетические инструменты, доступные в то время, было обнаружено, что вирус H1N1 1977 года тесно связан с вирусами человеческого гриппа H1N1, циркулировавшими в 1949–1950 годах, но не с теми, которые циркулировали раньше или позже.
…
Только начиная с 2009–2010 гг. в научных работах стало прямо указываться, что появление гриппа H1N1 в 1977 году было лабораторной утечкой: «Наиболее известный случай утечки лабораторного штамма — это вновь возникший вирус гриппа A H1N1, который впервые был обнаружен в Китае в мае 1977 года и вскоре после этого в России».
…
Предположение о том, что утечка 1977 г. могла быть связана с исследованиями вакцин против H1N1, подтверждается наблюдением, что в начальных вспышках в Китае девять из десяти вирусных изолятов выражали «температурную чувствительность» (Kung 1978). Чувствительность к температуре, как правило, необычная черта, но в 1970-х годах она была (и всё ещё остается) фундаментальной чертой для создания ослабленных живых вакцин против гриппа. Чувствительность к температуре обычно возникает только после серии существенных лабораторных манипуляций и отборов.
Интересно, что дальнейшие исследования показали, что циркулирующие штаммы в 1977–78 гг. часто состояли из смешанных температурно-чувствительных и нормальных компонентов, и что чувствительность к температуре, по-видимому, быстро исчезла из штамма H1N1 после 1978 г. Утечка штамма H1N1, проходящего лабораторное ослабление путём создания чувствительных к температуре мутантов, могла обеспечить как раз такую смешанную популяцию. В 1976–77 годах лабораторный персонал младше 20 лет не был знаком со штаммами гриппа H1N1 до 1957, поэтому мог быть подвержен инфицированию в лаборатории. Низкая тяжесть пандемии 1977 года может быть отчасти обусловлена чувствительностью вируса к температуре, что ограничивает репликацию вируса в легочных тканях.
Исходный текстHuman influenza H1N1 viruses appeared with the 1918 pandemic, and persisted, slowing accumulating small changes in its genome (with a major change in 1947), until the H2N2 “Asian” flu appeared in 1957, causing a worldwide pandemic. H1N1 influenza virus then apparently became extinct, and was not isolated for 20 years. In 1969 the “Hong Kong” H3N2 virus replaced the H2N2 virus, and is still circulating.
In September 1977 an H1N1 influenza virus was isolated from human infections in the Far East region of the Soviet Union, and in early 1978 the Chinese reported they had isolated H1N1 virus in May of 1977 in northeast China adjacent to the Soviet outbreak. Using the early genetic tools available at the time, the 1977 H1N1 virus was found to be closely related to H1N1 human influenza viruses circulating in 1949–1950, but not to those circulating earlier or later.
…
Only since 2009–2010 did major papers begin to state directly the 1977 emergence of H1N1 influenza was a laboratory related release: “The most famous case of a released laboratory strain is the re-emergent H1N1 influenza A virus which was first observed in China in May of 1977 and in Russia shortly thereafter.”
…
The speculation that the 1977 release may have been related to H1N1 vaccine research is supported by the observation that in the initial outbreaks in China, nine of the ten viral isolates expressed “temperature sensitivity” (Kung 1978). Temperature sensitivity normally an uncommon trait, but one that was in the 1970s (and still is) a fundamental trait for making live attenuated influenza vaccines. Temperature sensitivity generally occurs only after a series of substantial laboratory manipulations and selections.
Interestingly, further investigation indicated the circulating strains in 1977–78 were often comprised of mixed temperature-sensitive and normal components, and that temperature sensitivity apparently disappeared from the post-1978 H1N1 lineage rapidly. Escape of a mid-protocol population of H1N1 virus undergoing laboratory selection for temperature sensitive mutants would provide such a mixed population. In 1976–77 laboratory personnel in their late teens or early 20s would not have been exposed to pre-1957 H1N1 influenza viruses, and been susceptible to laboratory infections. The low severity of the 1977 pandemic might be in part due to the temperature sensitivity of the virus, a trait that limits virus replication in pulmonary tissues.
Могло ли что-то подобное стать причиной коронавирусной пандемии 2019-го? Почему нет? Тут возможно несколько вариантов — от разработки потенциальной вакцины до просто исследовательских работ по лабораторной рекомбинации летучемышиного и панголиньего вирусов. Какой-нибудь особо амбициозный исследователь мог даже просто по личной инициативе решить объединить две «модные темы» — добавление фуринового сайта и пересадку RBM из штамма одного вида (панголины) в другой (летучие мыши), чтобы потом, подтвердив повышенную вирулентность новой химерной конструкции, выпустить очередную грозную статью о том, какая опасность поджидает человечество в юньнаньских пещерах или на мокрых рынках. А если ещё и вакцину против такого опасного штамма удалось бы превентивно создать, то почёт и уважение такому исследователю были бы гарантированы.
Утверждаю ли я, что так всё и было? Конечно же, нет. Потому что на сегодняшний день доказательства такого сценария отсутствуют. Есть только череда странных совпадений — например, что вспышка юньнаньского коронавируса произошла за тысячи километров от Юньнани именно на том рынке, который ближе всего к Уханьскому институту вирусологии. А может, и не на рынке, так как из первых 4-х заболевших пациентов, трое на рынке не бывали. Ну и совпадения по структурным особенностям генома вируса, которые напоминают те манипуляции, которые вирусологи не раз проводили с такими вирусами в лаборатории. Но совпадения — это ещё не доказательства.
Более того, совпадения случаются, и, конечно же, в природе такой штамм тоже вполне мог возникнуть. Пока не очень ясно как именно — для этого летучемышиный и панголиний штаммы должны были встретиться в одной клетке (клетке чьего-то организма, а не металлической), причём в Ухане, так как вспышка произошла именно там (иначе мы бы видели другие очаги Ковида по пути следования первого зооносителя в Ухань). С учётом того, что летучие мыши на уханьском рынке не продавались, и вообще в это время года в спячке, такой вариант всё ещё остаётся неразгаданной тайной.
Кстати, совсем недавно появилась новость, что в 2018-ом году американские специалисты проводили инспекцию Уханьского института вирусологи, и даже общались с Ши Чжэнли. Результатом их «экскурсии» стали две дипломатических телеграммы в Вашингтон, в которых они отметили ряд слабых мест в обеспечении безопасности лаборатории:
Источники, знакомые с телеграммами, сказали, что они должны были поднять тревогу по поводу серьезных проблем безопасности в лаборатории WIV, особенно в отношении её работы с коронавирусами летучих мышей. Сотрудники посольства призывали к тому, чтобы США уделяли больше внимания этой лаборатории и оказывали ей дополнительную поддержку.Забавно, что уханьцам помогала та самая техасская лаборатория в Галвестоне, которая в своё время сама потеряла пробирку с венесуэльским вирусом. Причём помогала она не только на словах, там действительно проходили обучение уханьские специалисты, о чём даже писал «уханьский вестник» (правда, сейчас публикацию с сайта удалили, но на вебархиве она пока доступна):
…
«Во время взаимодействия с учеными из лаборатории WIV они отметили, что в новой лаборатории существует серьезная нехватка надлежащим образом подготовленных технических специалистов и исследователей, необходимых для безопасной эксплуатации этой лаборатории с высоким уровнем защиты», — говорится в телеграмме от 19 января 2018 года, которую подготовили два сотрудника отдела охраны окружающей среды, науки и здравоохранения посольства, лично встречавшиеся с учеными из WIV. (Государственный департамент отказался комментировать эту и другие детали истории.)
Китайские исследователи в WIV получали помощь от Галвестонской Национальной лаборатории в Медицинском отделении Университета Техаса и других организаций в США, но китайцы обратились за дополнительной помощью. В телеграммах утверждается, что Соединенные Штаты должны оказать дальнейшую поддержку лаборатории в Ухане, главным образом потому, что их исследование коронавирусов летучих мышей было важным, но также и опасным.
Исходный текстSources familiar with the cables said they were meant to sound an alarm about the grave safety concerns at the WIV lab, especially regarding its work with bat coronaviruses. The embassy officials were calling for more U.S. attention to this lab and more support for it, to help it fix its problems.
…
“During interactions with scientists at the WIV laboratory, they noted the new lab has a serious shortage of appropriately trained technicians and investigators needed to safely operate this high-containment laboratory,” states the Jan. 19, 2018, cable, which was drafted by two officials from the embassy’s environment, science and health sections who met with the WIV scientists. (The State Department declined to comment on this and other details of the story.)
The Chinese researchers at WIV were receiving assistance from the Galveston National Laboratory at the University of Texas Medical Branch and other U.S. organizations, but the Chinese requested additional help. The cables argued that the United States should give the Wuhan lab further support, mainly because its research on bat coronaviruses was important but also dangerous.
Последний штрих к семейному портрету лабораторных утечек: в ноябре 2019-го в Ланьчжоу сразу в двух исследовательских центрах произошла вспышка бруцеллёза (бактериальной инфекции), поразившая более 100 работавших там исследователей.
Возможные следы рукотворности
Засим оставим вирусологов в покое и обратим наш взор опять на сам вирус. Есть ли в нём какие-то явные признаки рукотворности? Для начала пару слов о том, что значит «явные». Понятно, что в природе могут произойти любые мутации совершенно случайно. Даже если бы врезка, создавшая фуриновый сайт в CoV2 была не “PRRA”, а “MADEINWVHANPRRA”, то всё равно оставался бы ненулевой шанс, что она могла возникнуть случайно. Но для нас, да и для любого суда, думаю, этого было бы достаточно, чтобы доказать рукотворное происхождение beyond a reasonable doubt.
Главная проблема с такими доказательствами состоит в том, что даже в рукотворном вирусе их попросту может не быть. Грубо говоря, хороший генный инженер может создать синтетический вирус «идентичный натуральному». Более того, часто исследователи намеренно привносят в свои конструкции какие-нибудь синонимичные мутации для того, чтобы потом можно было различить их штамм и природный. Но если создатель вируса эти маркеры рукотворности сам не раскрывает, отличить их от природных мутаций невозможно.
Но иногда следы могут и оставаться, особенно если создатели не пытаются скрыть рукотворность своей конструкции. В первую очередь, речь о местах разрезов ДНК (напомню, что манипуляции с РНК-вирусами проводятся именно в комплементарных им ДНК-конструкциях), необходимых создателям для сшивания разных сегментов генома или для вырезания старых и вставки новых участков. Ведь ДНК можно разрезать не в произвольных местах (Криспер не в счёт), а только там, где последовательность нуклеотидов (обычно 4–6 «букв») совпадает с той последовательностью, которую распознаёт та или иная рестриктаза, то есть фермент, расщепляющий цепочки нуклеотидов. При этом такой анализ осложняет то, что существуют сотни различных типов рестриктаз, используемых в генной инженерии. Но давайте попробуем провести такой анализ для CoV2.
Для начала — пример работы группы Барика от 2008 года, где они взяли Bat-SCoV и заменили RBD в его шиповидном белке на RBD из человеческого SARS-а. Вот как они описывают создание своей химеры:
Схематическое представление вариантов SARS-CoV и Bat-SCoV.Как видим, сначала группа Барика создала синтетический клон летучемышиного Bat-SCoV, причём «по лекалам» уже созданного ими ранее синтетического клона SARS-CoV. То есть для летучемышиного клона они использовали те же 6 сегментов с теми же сайтами рестриктаз, которые ранее они использовали для SARS-CoV, что позволило им менять местами сегменты вируса между разными штаммами как куски Лего. Вот подробное описание создания химерного мутанта:
(A) Схематическое представление геномов SARS-CoV и Bat-SCoV (инвентарный номер GenBank FJ211859) и системы обратной генетики. (Вверху) стрелки указывают сайты процессинга вирусной протеазы nsp внутри полипротеина ORF1ab (открытые стрелки, папаин-подобная протеаза; закрашенные стрелки, nsp5 [3C-подобная протеаза]). Сразу ниже приведены фрагменты, использованные в системе обратной генетики, обозначенные от A до F. Фрагменты, синтезированные для создания Bat-SCoV, точно воспроизводят соединения фрагментов SARS-CoV, за исключением того, что Bat-SCoV имеет 2 фрагмента, Bat-E1 и Bat-E2, которые соответствуют фрагменту SARS-E.
Исходный текстSchematic representation of SARS-CoV and Bat-SCoV variants.
(A) Schematic representation of SARS-CoV and Bat-SCoV (GenBank accession no. FJ211859) genomes and reverse genetics system. (Top) Arrowheads indicate nsp processing sites within the ORF1ab polyprotein (open arrowheads, papain-like proteinase mediated; filled arrowheads, nsp5 [3C-like proteinase] mediated). Immediately below are the fragments used in the reverse genetics system, labeled A through F. The fragments synthesized to generate Bat-SCoV exactly recapitulate the fragment junctions of SARS-CoV with the exception that the Bat-SCoV has 2 fragments, Bat-E1 and Bat-E2, which correspond to the SARS-E fragment.
Вирусы, содержащие ПЦР-генерированные вставки в вирусный геном, были получены с использованием стратегии сборки SARS-CoV (24, 33, 53) со следующими модификациями. Вкратце, для вируса Bat-F кДНК полной длины конструировали путем лигирования продуктов рестрикции из фрагментов A-E SARS-CoV и фрагмента F Bat-SCoV, что требовало расщепления Bgl-NotI. Для Bat-SCoV и Bat-SRBD, Bat-SRBM и Bat-Hinge плазмиды, содержащие 7 фрагментов кДНК генома Bat-SCoV, были расщеплены с использованием BglI для Bat-A, Bat-B, Bat-C и Bat -D, BglI и AflII для Bat-E1 и Bat-E2 и BglI и NotI для Bat-F. Расщепленные, очищенные фрагменты одновременно лигировали вместе. Транскрипция осуществлялась с использованием набора m7Message mMachine (Ambion), затем РНК электропорировали в клетки Vero (24, 53).Все эти трёхбуквенные аббревиатуры (BglI, AflII,NotI и т.д.) в выделенном выше предложении и являются различными типами рестриктаз. Давайте посмотрим, будут ли заметны какие-то различия в сайтах рестриктаз в геноме (шиповидного белка) химеры в сравнении с геномом исходного SARS-CoV:
Исходный текстViruses containing PCR-generated insertions within the viral coding sequence were produced by using the SARS-CoV assembly strategy (24, 33, 53) with the following modifications. Briefly, for Bat-F virus, full-length cDNA was constructed by ligating restriction products from SARS-CoV fragments A–E and Bat-SCoV fragment F, which required a BglI-NotI digestion. For Bat-SCoV and Bat-SRBD, Bat-SRBM, and Bat-Hinge, plasmids containing the 7 cDNA fragments of the Bat-SCoV genome were digested by using BglI for Bat-A, Bat-B, Bat-C, and Bat-D, BglI and AflII for Bat-E1 and Bat-E2, and BglI and NotI for Bat-F. Digested, gel-purified fragments were simultaneously ligated together. Transcription was driven by using a T7 mMessage mMachine kit (Ambion), and RNA was electroporated into Vero cells (24, 53).
Как видно, сайты рестриктаз у химеры практически идентичны тем кускам исходных последовательностей в Bat-SCoV или SARS, откуда они были взяты. Единственные различия заметны в местах сшивания вставленного куска из SARS. Вот, например левый (5’-) край вставки:
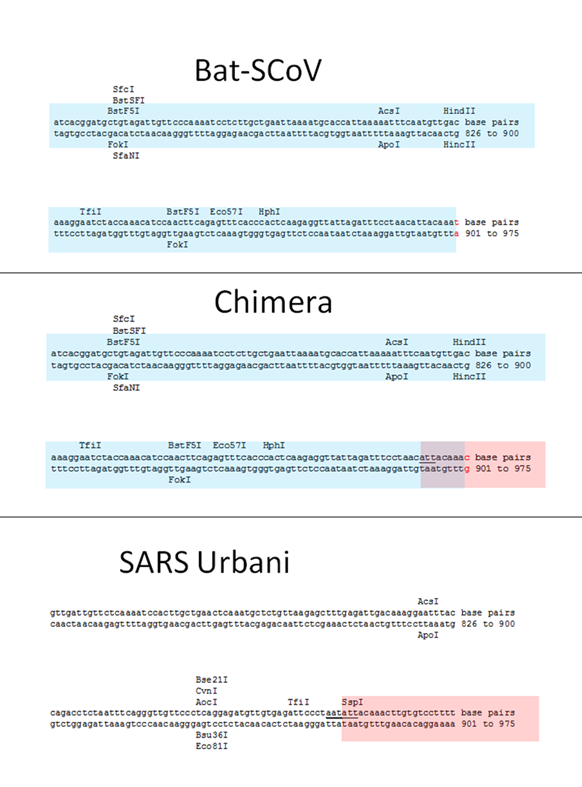
Здесь у Bat-SCoV и SARS оказалась общая идентичная область нуклеотидов (пересечение бирюзовой и розовой областей), и в месте сшивки двух последовательностей никаких новых сайтов рестриктаз нет, а наоборот пропал сайт SspI из SARS-а. А вот правый (3’-) край вставки:
Тут в месте склеивания наоборот остались все старые рестриктазные сайты, и даже появились новые, например, EcoRII. Если бы я не знал, что химерный геном является следствием рукотворных манипуляций, смог бы я это понять, глядя на эти 3 сиквенса? Вряд ли, даже если бы у меня и закрались какие-то подозрения, то точно не beyond a reasonable doubt. Быть может, специалистам в генной инженерии это было бы видно по каким-то другим признакам, утверждать не берусь — здесь буду рад любому ликбезу.
Но в любом случае, давайте сравним шиповидный белок у RaTG13, CoV2 и панголина-19. Вдруг там что-нибудь на нас как выскочит, как выпрыгнет!
Вот так выглядит RBD (выделен салатовым) и RBM (жёлтым) у всех троих:
Что тут интересного? Любопытно было видеть сайт EcoRI на 5’-крае RBM у всех троих (первый стык между салатовой и жёлтой областями) — весьма удобно. Интересно, насколько это распространённая фича у других штаммов? Беглый анализ показал, что наша троица — рекордсмены по количеству таких сайтов в геноме, у других летучемышиных штаммов их всего по 5:
Возвращаясь к анализу RBD, из особенностей CoV2 можно отметить новые сайты рестриктаз, выделенные красными прямоугольниками — они совпадают с уникальными мутациями в аминокислотной последовательности (тоже отмечены красными прямоугольниками на аминокислотных сиквенсах в крайней правой колонке). На всякий случай я выделил ещё несколько новых сайтов: голубые прямоугольники и зелёный прямоугольник, который находится в районе единственной аминокислоты, различающейся между RBM CoV2 и панголина-19.
Давайте ещё сравним у всех троих штаммов место врезки PRRA, создавшей в CoV2 фуриновый сайт:

Тут тоже появилось несколько новых сайтов (выделены голубым) по обе стороны от новой вставки. Могли ли они быть использованы для создания фуринового сайта? Теоретически да. Но вставку можно было сделать и используя имеющиеся сайты, или даже создав сегменты с новыми сайтами, которые затем соединить «бесшовно», то есть не создавая новых сайтов на стыке. Если помните, Барик ещё в 2002 году применил эту технологию для создания синтетического клона мышиного коронавируса:
Места соединения сайтов рестрикции, которые расположены на концах каждой кДНК, систематически удаляются во время сборки полного полноразмерного продукта кДНК, что позволяет проводить повторную сборку без внесения изменений нуклеотидов.А в 2003 повторил на синтетическом клоне SARS-CoV:
Исходный текстThe interconnecting restriction site junctions that are located at the ends of each cDNA are systematically removed during the assembly of the complete full-length cDNA product, allowing reassembly without the introduction of nucleotide changes.
Чтобы быстро собрать консенсусные клоны, мы использовали рестрикционные эндонуклеазы класса IIS, которые разрезают в асимметричных участках и оставляют асимметричные концы. Эти ферменты генерируют специфичные уникальные оверхенги, которые обеспечивают бесшовное лигирование двух кДНК с последующей потерей рестриктазного сайта.Сегодня технология жонглирования генными последовательностями уже настолько автоматизирована и поставлена на поток, что в китайской статье от октября 2019 года про врезку нового фуринового сайта в куриный коронавирус её описанию отведено лишь пару предложений:
Исходный текстTo rapidly assemble consensus clones, we used class IIS restriction endonucleases that cut at asymmetric sites and leave asymmetric ends. These enzymes generate strand-specific unique overhangs that allow the seamless ligation of two cDNAs with the concomitant loss of the restriction site.
2.2. Создание рекомбинантного вирусаНевозможно не восхищаться тем, до чего дошел прогресс! Вот описание вышеупомянутого набора для бесшовной сборки Seamless Assembly kit:
Рекомбинантный вирус rYN-S2/RRKR, содержащий шиповидный белок с фуриновым сайтом S2', был получен путем vaccinia рекомбинации, как описано ранее [20, 28]. Вкратце, плазмиду с фуриновым сайтом S ‘генерировали с использованием набора для бесшовной сборки Seamless Assembly kit (Invitrogen, Carlsbad, CA, USA) и трансфицировали в клетки CV-1, инфицированные вирусом vaccinia, содержащим геном YN-?S-GPT. Сайт Furin-S2 был введен в кДНК YN путем гомологичной рекомбинации с использованием системы временной доминантной селекции [25].
Исходный текст2.2. Generation of Recombinant Virus
Recombinant rYN-S2/RRKR virus containing an S protein with the furin-S2? site was generated by vaccinia recombination, as described previously [20,28]. Briefly, plasmid with the furin-S2? site was generated using the Seamless Assembly kit (Invitrogen, Carlsbad, CA, USA) and transfected into CV-1 cells infected by vaccinia virus containing the genome of YN-?S-GPT. Furin-S2’ site was introduced into the YN cDNA by homologous recombination using the transient dominant selection system [25].
Набор для бесшовного клонирования и сборки GeneArt позволяет одновременно и направленно клонировать от 1 до 4 фрагментов ПЦР, состоящих из любой последовательности, в любой линеаризованный вектор с помощью одной 30-минутной реакции при комнатной температуре. Набор содержит всё необходимое для сборки фрагментов ДНК и их трансформации в E.coli для отбора и роста рекомбинантных векторов.До 4-х фрагментов ДНК можно склеить в нужной последовательности за каких-то полчаса, причём без головной боли с рестриктазами или лигацией. И загрузить своё творение в E. coli для размножения полученной конструкции.
• Скорость и простота — клонируйте до 4 фрагментов ДНК с выбранной последовательностью одновременно в одном векторе (до 13 Кб); не требуются сайты рестрикции, лигирования или рекомбинации
• Точность и эффективность — предназначен для клонирования того, что вы хотите, где вы хотите, в нужной ориентации и достижения до 90% правильных клонов без лишних последовательностей.
• Гибкость вектора — используйте наш линейный вектор или вектор по вашему выбору
• Бесплатные инструменты — Создавайте ДНК-олиго и многое другое с помощью нашего бесплатного веб-интерфейса, который шаг за шагом проведет вас через ваш проект
• Разнообразные приложения — Оптимизируйте многие методы синтетической биологии и молекулярной биологии с помощью быстрой комбинации, добавления, удаления или обмена сегментов ДНК.
Исходный текстThe GeneArt Seamless Cloning and Assembly Kit enables the simultaneous and directional cloning of 1 to 4 PCR fragments, consisting of any sequence, into any linearized vector, in a single 30-minute room temperature reaction. The kit contains everything required for the assembly of DNA fragments, and their transformation into E. coli for selection and growth of recombinant vectors.
• Speed and Ease?—?Clone up to 4 DNA fragments, with sequence of your choice, simultaneously in a single vector (up to 13 Kb); no restriction digestion, ligation or recombination sites required
• Precision and Efficiency — Designed to let you clone what you want, where you want, in the orientation you want, and achieve up to 90% correct clones with no extra sequences left behind
• Vector Flexibility — Use our linear vector or a vector of your choice
• Free Tools — Design DNA oligos and more with our free web-based interface that walks you step-by-step through your project
• Diverse Applications — Streamline many synthetic biology and molecular biology techniques through the rapid combination, addition, deletion, or exchange of DNA segments
Подводя черту под анализом рестриктазных сайтов, необходимо признать, что никаких однозначных выводов по его итогам сделать нельзя. Разве что в очередной раз можно было убедиться, что не только CoV2 уникален, но и сам RaTG13 очень необычный товарищ, и что стоит дальше изучать его биографию.
Кодоновые предпочтения
В этих целях я ещё решил взглянуть на codon usage bias, чтобы посмотреть на какие штаммы других вирусов похожи CoV2 и RaTG13. Не секрет, что вирусы склонны подстраиваться по своей кодоновой сигнатуре под предпочтения их хозяев, поэтому я ожидал увидеть у RaTG13 схожий паттерн с другими лечучемышиными вирусами, а также надеялся увидеть отличие от панголиньих штаммов.
Вот SARS-CoV, например, очень похож на Rs3367 и RsSCH014, как и можно было ожидать:
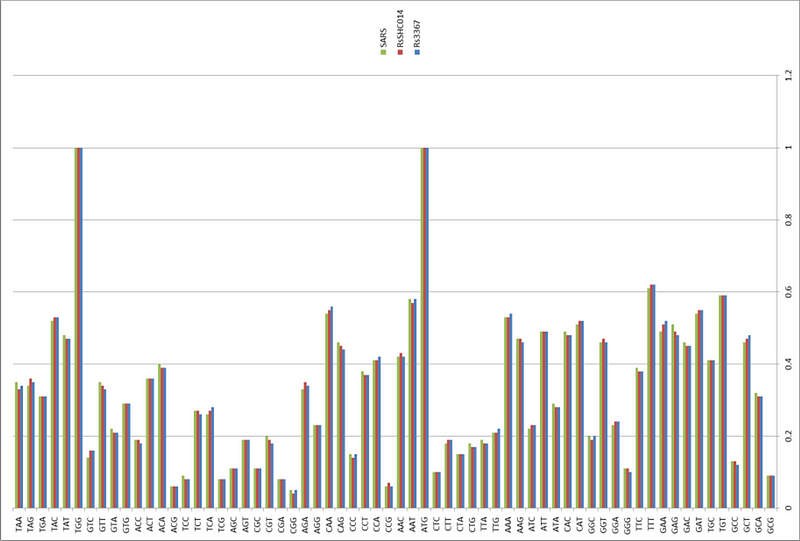
Между собой, кстати, SARS, MERS и CoV2 различаются:
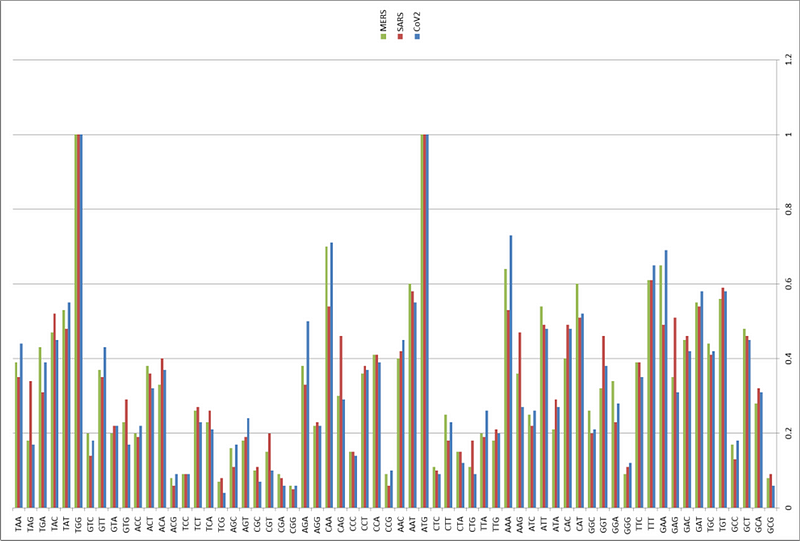
RaTG13 похож на CoV2, что тоже вполне ожидаемо:
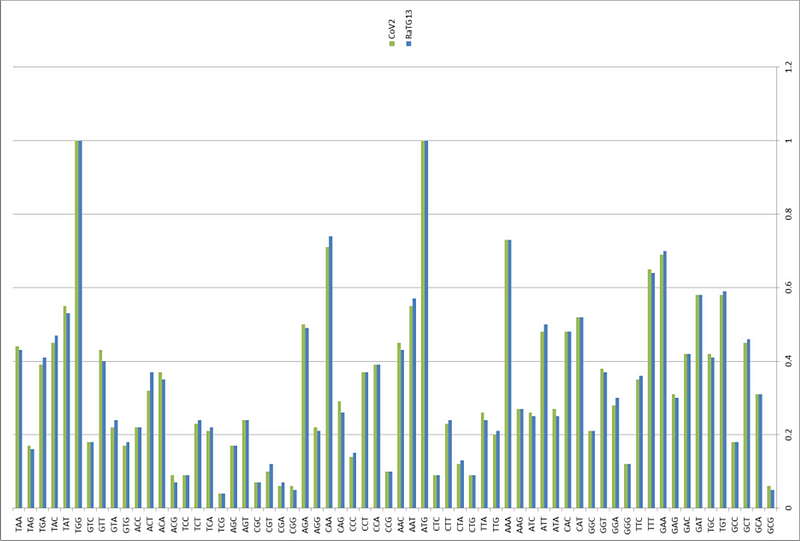
А вот на панголиньи штаммы RaTG13 действительно не супер похож, да и между собой панголиньи штаммы не то чтобы идентичны:
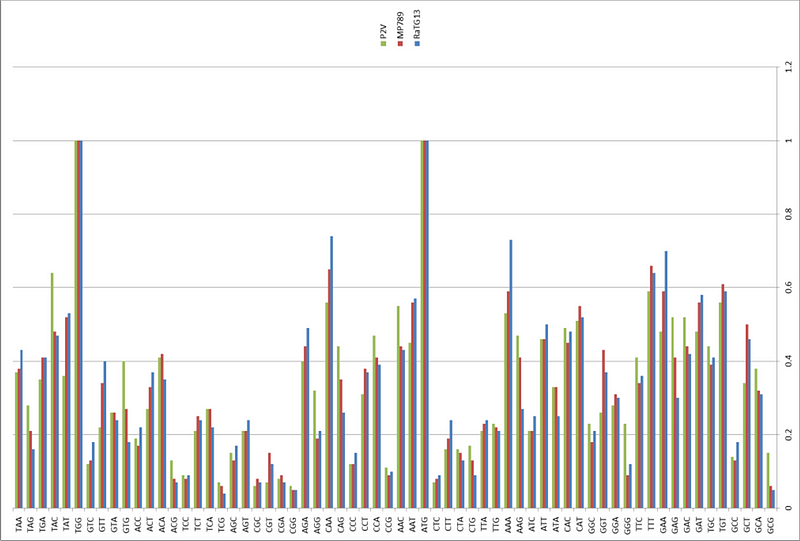
На ZXC21 и ZC45 тоже не сильно похож:
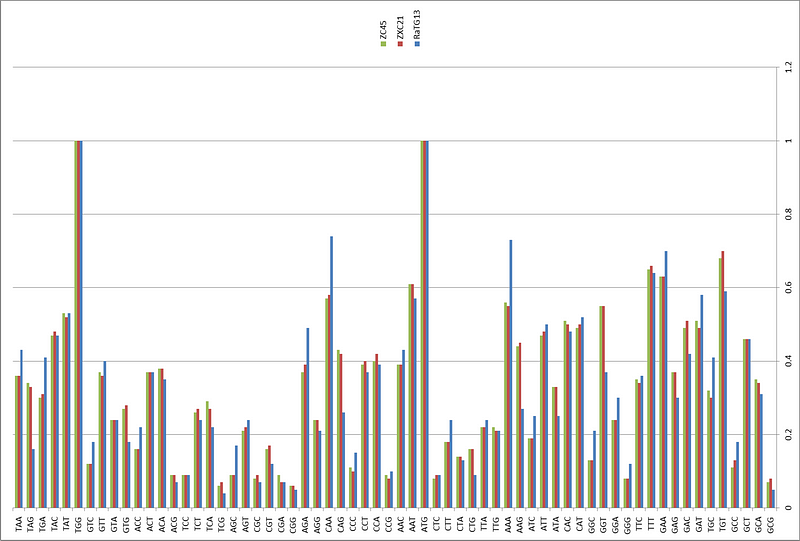
Из юньнаньских штаммов RaTG13 от Rs3367 и RsSCH014 довольно далёк, а ближе всего к LYRa11, но тоже с заметными различиями:
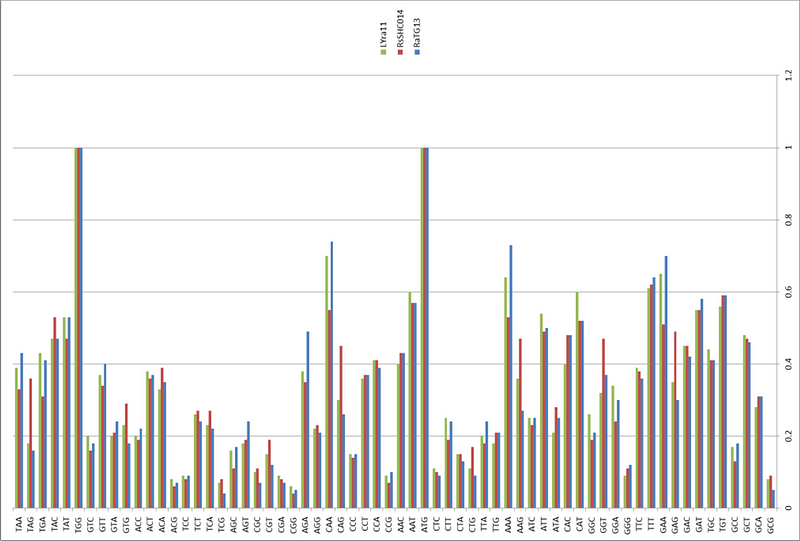
В общем, как всегда, RaTG13 и CoV2 стоят как-то обособленно. Ещё меня заинтриговал AAA кодон — он у них используется намного чаще, чем у соплеменников:
Вероятно, это просто очередное совпадение, но схожая пропорция между AAA и AAG наблюдается у E. coli. Может ли меняться кодоновая сигнатура у cDNA по мере её длительной культивации в клеточной культуре? В теории, да, но я пока глубоко эту тему не копал.
Что ж, кодоновый анализ тоже не выявил каких-то явных признаков рукотворности, но в очередной раз подтвердил уникальность CoV2 и RaTG13. Что у нас есть в сухом отстатке? Пока что лишь некий набор странных совпадений, который, как любят выражаться учёные, taken together, то есть, в совокупности, заставляет очень сильно задуматься. И уж точно не позволяет отвергнуть гипотезу о рукотворной природе CoV2.
А как же опровержение рукотворности в Nature?
Как не позволяет? А почему авторам той статьи в Nature позволяет? На самом деле никакого опровержения рукотворности в той статье нет. Есть только громкое «нам так не кажется», базирующееся на весьма зыбком фундаменте. Сами посудите — вот основные тезисы авторов в поддержку нерукотворности:
Хотя приведенный выше анализ предполагает, что SARS-CoV-2 может связываться с человеческим ACE2 с высокой аффинностью, вычислительный анализ предсказывает, что его взаимодействие не является идеальным, и что его последовательность RBD отличается от показанной оптимальной последовательности для SARS-CoV [первый SARS — Ю.Д.] для связывания с рецептором. Таким образом, высокоаффинное связывание шиповидного белка SARS-CoV-2 с человеческим ACE2, скорее всего, является результатом естественного отбора на человеческом или подобном человеку ACE2, который позволил возникнуть другому оптимальному решению для связывания. Это убедительное доказательство того, что SARS-CoV-2 не является продуктом целенаправленных манипуляций.Эта цитата в статье приведена прямо под диаграммой, на которой показаны идентичные RBM у CoV2 и панголина-19. Погодите, тогда при чём тут «компьютерное моделирование»? Наиболее вероятный сценарий рукотворности — перенос RBM из штамма одного животного в штамм другого — что вирусологи уже делали многократно. Поэтому логическая цепочка авторов не выдерживает никакой критики: «на компьютере можно было и круче вирус сдизайнить, значит CoV2 — результат естественного отбора. О, значит это и есть весомое доказательство того, что CoV2 нерукотворен!» Видимо, плохо с логикой у авторов. Дальнейшие их тезисы это подтверждают:
Исходный текстWhile the analyses above suggest that SARS-CoV-2 may bind human ACE2 with high affinity, computational analyses predict that the interaction is not ideal and that the RBD sequence is different from those shown in SARS-CoV to be optimal for receptor binding. Thus, the high-affinity binding of the SARS-CoV-2 spike protein to human ACE2 is most likely the result of natural selection on a human or human-like ACE2 that permits another optimal binding solution to arise. This is strong evidence that SARS-CoV-2 is not the product of purposeful manipulation.
Кроме того, если бы была проведена генетическая манипуляция, вероятно, была бы использована одна из нескольких обратных генетических систем, доступных для бета-коронавирусов. Тем не менее, генетические данные неопровержимо показывают, что SARS-CoV-2 не получен из какой-либо ранее использованной вирусной основы.Опять та же логическая ошибка, завуалированная громкими оборотами: «генетический анализ неопровержимо доказывает, что CoV2 абсолютно точно не был создан на основе ранее известных вирусов!» Ну спасибо, Капитан Очевидность. И что, неужели создатели вируса не могли сделать cDNA backbone из ранее не публиковавшегося штамма вируса — например, из того же RaTG13? Да легко. Также им не составило бы труда затем туда вставить панголиний RBM и фуриновый сайт. Вирусологи этим занимаются 20 лет, а современный инструментарий генной инженерии делает такие манипуляции доступными даже студенту.
Исходный текстFurthermore, if genetic manipulation had been performed, one of the several reverse-genetic systems available for betacoronaviruses would probably have been used. However, the genetic data irrefutably show that SARS-CoV-2 is not derived from any previously used virus backbone.
Насчёт возникновения фуринового сайта в клеточной культуре авторы тоже высказывают странные тезисы:
Приобретение как фуринового сайта расщепления, так и предсказанных O-связанных гликанов также противоречит сценариям возникновения в клеточной культуре. Новые фуриновые сайты расщепления наблюдались только после длительного пассирования вируса птичьего гриппа с низкой патогенностью in vitro или in vivo. Кроме того, гипотетическая генерация SARS-CoV-2 в клеточной культуре или пассировании в животных потребовала бы предварительного выделения вируса-предшественника с очень высоким генетическим сходством, которое не было описано. Последующее образование фуринового сайта потребовало бы повторного пассирования в клеточной культуре или у животных с рецепторами ACE2, сходными с таковыми у людей, но такая работа также ранее не была описана.Во-первых, сами же авторы ранее приводят ссылки на работы, где фуриновый сайт возникал по мере культивирования вирусов в клетках. А во-вторых, что значит, не описан близкий штамм вируса — а как же RaTG13? Если в нём синтетически заменить RBM на панголиний, а затем культивировать химерный штамм в клеточной культуре, то фуриновый сайт вполне мог возникнуть и таким образом, и вдобавок, таким же путём новый штамм мог приобрести и другие мутации, отличающие CoV2 от RaTG13 и панголина-19.
Исходный текстThe acquisition of both the polybasic cleavage site and predicted O-linked glycans also argues against culture-based scenarios. New polybasic cleavage sites have been observed only after prolonged passage of low-pathogenicity avian influenza virus in vitro or in vivo. Furthermore, a hypothetical generation of SARS-CoV-2 by cell culture or animal passage would have required prior isolation of a progenitor virus with very high genetic similarity, which has not been described. Subsequent generation of a polybasic cleavage site would have then required repeated passage in cell culture or animals with ACE2 receptors similar to those of humans, but such work has also not previously been described.
Но в плане именно рукотворного варианта возникновения фуринового сайта мне более вероятным видится вариант с целенаправленной врезкой — как в другой китайской работе от октября 2019 года с куриным коронавирусом. А уже затем созданный штамм мог приобрести новые мутации по мере его культивирования in vitro или in vivo — как мышиный штамм MA15 в 2007 году, например.
Ши Чжэнли-2020
Пока я писал эту статью, вышла работа большого коллектива китайских вирусологов, включая Ши Чжэнли, в которой они ещё в феврале протестировали против CoV2 свою многолетнюю разработку от многих видов коронавирусов — некий пептид, который призван блокировать слияние шиповидного белка с клеточной мембраной. Авторы, конечно же, упоминают новый фуриновый сайт CoV2, и предполагают, что он может играть важную роль в гораздо более эффективном проникновении CoV2 в клетку:
В этом исследовании мы показали, что SARS-CoV-2 демонстрирует гораздо более высокую способность слияния с мембраной, чем SARS-CoV, что позволяет предположить, что механизм слияния SARS-CoV-2 является важной мишенью для разработки ингибиторов слияния коронавируса.В этом контексте становится интересно, а не проводили ли авторы ранее какие-то эксперименты по тому, как на эффективность их пептида может влиять добавление новых фуриновых сайтов в различные коронавирусы? Но не будем строить лишние догадки, позволим времени расставить всё по своим местам.
…
Как правило, в β-В коронавирусах отсутствует фуриновый сайт S1/S2, а их шиповидные белки в нативном состоянии не расщеплены. Например, SARS-CoV проникает в клетку главным образом через эндосомальный путь слияния с мембраной, где его шиповидный белок расщепляется эндосомным катепсином L и активируется. Приобретение фуринового сайта S1/S2 может значительно увеличить способность шиповидного белка SARS-CoV проникать через клеточную мембрану.
Исходный текстIn this study, we have shown that SARS-CoV-2 exhibits much higher capacity of membrane fusion than SARS-CoV, suggesting that the fusion machinery of SARS-CoV-2 is an important target for development of coronavirus fusion inhibitors.
…
Generally, β-B coronaviruses lack the S1/S2 furin-recognition site, and their S proteins are uncleaved in the native state. For example, SARS-CoV enters into the cell mainly via the endosomal membrane fusion pathway where its S protein is cleaved by endosomal cathepsin L and activated. Inducing the S1/S2 furin-recognition site could significantly increase the capacity of SARS-CoV S protein to mediate cellular membrane surface infection.
Кстати, Барик, видимо, решил не отставать от Ши Чжэнли, и тоже присоединился к гонке по поиску средств от CoV2. Как я понял, он и соавторы взяли уже имевшиеся у них данные об эффективности своего нуклеозидного аналога (β-D-N4-hydroxycytidine, NHC) против SARS-CoV и MERS, добавили in vitro данные по CoV2, и отправили в печать. Нуклеозидные аналоги (как, например, знаменитый ремдесивир) — это принципиально другой подход, чем у Ши Чжэнли и соавторов: тут авторы пытаются помешать репликации вируса, подсовывая ему «дефективные» буквы генетического алфавита, а Ши Чжэнли и соавторы пытаются не дать вирусу проникнуть в клетку. Теоретически, эти подходы можно будет комбинировать.
This is the end, beautiful friend
Ох, надеюсь до этого момента хоть кто-то дочитает. Простите, если утомил вас. Сам в шоке: кроличья нора оказалась целым подземным царством. Ещё надеюсь, что вам было интересно погрузиться в мир вирусологии и непредвзято рассмотреть гипотезу рукотворной природы CoV2. На мой взгляд, приведённый мной массив данных, в совокупности, не позволяет отвергнуть эту гипотезу.
На всякий случаю, поясню: это НЕ означает, что CoV2 точно был синтезирован в лаборатории. Да, чисто технически, создать такой штамм современному вирусологу не составило бы труда. Но прямых доказательств тому, что кто-либо это сделал, нет. А странные совпадения пока не могут служить даже косвенными доказательствами.
Но и обратная гипотеза об исключительно натуральной природе вируса тоже ещё вескими доказательствами не подкреплена. Пока не обнаружено промежуточных предков между RaTG13, панголином-19 и CoV2, в которых бы можно было проследить ту селективную рекомбинацию, которую мы наблюдаем у CoV2, вопрос о его происхождении остаётся открытым. Пожалуй, лучше самого Ральфа Барика на эту тему не выскажется никто:
Какие животные являются зоонозными носителями SARS-CoV-2?Такие дела.
Такие животные пока не найдены. Есть сведения, что панголины потенциально могут быть промежуточным хозяином, но вирусы панголина лишь на 88–98% идентичны SARS-CoV-2. Для сравнения, штаммы цивет и енотовидных коронавирусов первого SARS были на 99,8% идентичны штаммам SARS-CoV 2003 года. Другими словами, мы говорим о нескольких мутациях между штаммами цивет, енотовидных и людей в 2003 году. Панголиньи [штаммы CoV2] имеют более 3000 нуклеотидных изменений, поэтому панголины никоим образом не могут быть резервуарным видом. Абсолютно без шансов.
Исходный текстWhat is the reservoir species of SARS-CoV-2?
They have not identified the actual reservoir species. Reports show that pangolins are potentially the intermediate host, but pangolin viruses are 88–98% identical to SARS-CoV-2. In comparison, civet and racoon dog strains of SARS coronaviruses were 99.8% identical to SARS-CoV from 2003. In other words, we are talking about a handful of mutations between civet strains, racoon dog strains and human strains in 2003. Pangolin [strains of CoV2] have over 3000 nucleotide changes, no way they are the reservoir species. Absolutely no chance.
****************************************************************************
UPD: о 4% разницы между геномами RaTG13 и Cov2
Некоторые критики лабораторной гипотезы утверждают, что наблюдаемое ~ 4% генетическое различие между RaTG13 и CoV2 слишком велико, чтобы это могло произойти в лаборатории, если сам RaTG13 использовался в качестве основы. Наблюдаемые скорости мутаций для РНК-вирусов варьируются в широких пределах — от 10-6 до 10-4 нуклеотидов на репликацию in vitro, а у людей CoV2, по-видимому, мутирует со скоростью 25 мутаций в год. Таким образом, по логике критиков, потребовались бы годы, если не десятилетия, чтобы эти два штамма разошлись на 4%. Несмотря на то, что это резонное возражение, я вижу несколько проблем с ним.
Во-первых, скорости мутаций in vitro намного выше чем in vivo, поскольку вы можете пассировать клетки гораздо чаще, чем заражать новых животных. Как показали эксперименты с SARS и MERS in vitro, существенные мутации могут наблюдаться уже после нескольких пассажей. Например, в статье 2004 года сообщалось, что после 600 пассажей уже наблюдалось 2,1% различий в геномных последовательностях шиповидный белков между исходным штаммом и его потомством:
Более того, в присутствии некоторых противовирусных препаратов, например, тех же нуклеозидных аналогов (рибавирина или ремдесивира), частота мутаций в РНК-вирусах может увеличиваться втрое:
Мы получили оценку частоты спонтанных мутаций ок. 10-4 замены на сайт или ниже, что находится в пределах общепринятого диапазона для РНК-вирусов. Примерно трехкратное увеличение частоты мутаций и значительное смещение мутационного спектра наблюдались в образцах пациентов, получавших лечение интерфероном и рибавирином в течение 6 месяцев. Этот результат согласуется с известным мутагенным действием рибавирина in vitro и предполагает, что противовирусный эффект лечения рибавирином и интерфероном, по крайней мере, частично проявляется в результате летального мутагенеза.Таким образом, если лабораторный предок CoV2 тестировался для оценки того, как его мутагенез может менять эффективность потенциальных вакцин или противовирусных препаратов против него, он вполне мог достаточно быстро накопить существенные различия.
Исходный текстWe obtained an estimate of the spontaneous mutation rate of ca. 10-4 substitutions per site or lower, a value within the typically accepted range for RNA viruses. A roughly threefold increase in mutation rate and a significant shift in mutation spectrum were observed in samples from patients undergoing 6 months of interferon plus ribavirin treatment. This result is consistent with the known in vitro mutagenic effect of ribavirin and suggests that the antiviral effect of ribavirin plus interferon treatment is at least partly exerted through lethal mutagenesis.
Но, возможно, самая большая проблема с аргументом о 4% -ной разнице заключается в том, что он основан на том, что штамм RaTG13 является именно тем, чем его объявил WIV, и что других, более близких штаммов в их коллекции нет. Если мы хотим непредвзято рассмотреть гипотезу о лабораторной утечке, мы должны признать, что не можем полностью доверять данным от той самой лаборатории, которая подозревается в утечке. Если утечка все-таки произошла, как предполагает гипотеза, то WIV уже пытается ее скрыть, и предоставляемые ими данные вполне могут следовать той же цели.
Ещё раз хочу подчеркнуть, что я не утверждаю со 100%-ой уверенностью, что именно это произошло. Всё, что я говорю, это то, что, несмотря на заявления некоторых уважаемых учёных об обратном, утечка всё же могла произойти, и нам нужно гораздо больше доказательств, прежде чем мы сможем прийти к окончательному выводу. Но — и это важный момент — даже если CoV2 действительно был случайной утечкой, сами ученые в этом не виноваты, так как они работали в рамках общепринятых международных законов, и ничего криминального в том, что они делали не было, и нет. У меня нет никакой антипатии к самим вирусологам, наоборот, я восхищаюсь тем, каких высот достигла фундаментальная биология и генная инженерия. А вот к тем, кто, если утечка произошла, пытается её скрыть, совсем другой разговор.
Кстати, есть кое-что, что могло бы помочь поверить утверждениям WIV о природе RaTG13 – если они согласятся передать независимым лабораториям свои юньнаньские образцы от 2013 года, из которых Ши Чжэнли извлекла RaTG13. У них они всё ещё должны быть, если они в 2020 году повторно из них секвенировали полный геном RaTG13.
****************************************************************************
UPD2: RaTG13 — тот же штамм, что и RaBtCoV/4991?
После того, как я опубликовал этот пост, мне указали на препринт, в котором утверждается, что RaTG13 на самом деле может являться штаммом RaBtCoV/4991 (KP876546), об обнаружении которого в 2013 году в заброшенной Юньнаньской шахте группа Ши Чжэнли сообщала в 2016 году. Есть несколько причин так считать. Прежде всего, единственная опубликованная последовательность из RaBtCoV/4991 на 100% идентична последовательности RaTG13 на уровне нуклеотидов, хоть это всего лишь и 370 нуклеотидов из гена RdRp:
Во-вторых, данные о сборе материала, из которых были выделены эти штаммы, практически идентичны: оба были собраны в июле 2013 года из мазка летучих мышей R. affinis:
Образец с RaBtCoV/4991 был собран на шахте, расположенной в округе Мудзян, который находится под юрисдикцией города Пуэр:
Автономный округ Мудзян является автономным округом под юрисдикцией города Пуэр, на юге провинции Юньнань, Китай. WikipediaА город Пуэр указан как место сбора RaTG13 в базе данных GISAID, что вполне может быть приблизительным указанием местоположения шахты в Мудзян.
Странно, что в своей статье от 2020 года о RaTG13 Ши Чжэнли не упоминает RaBtCoV/4991 и не цитирует свою статью 2016 года о его открытии, в которой она указана как «разработавшая и координировавшая исследование». При этом, вряд ли RaBtCoV/4991 был совсем забыт, так как он упоминается в статье группы Ши Чжэнли от 2019 года, где он включен в филогенетическое древо других коронавирусов:

Я сомневаюсь, что место RaBtCoV/4991 в этом дереве было определено исключительно на основе фрагмента в 370 нуклеотидов, поэтому я думаю, что к началу 2019 года группа Ши Чжэнли уже секвенировала его геном.
Кстати, интересно, что геномы панголина-2017 и панголина-2019 также очень близки к RaTG13 и CoV2 в этом участке гена RdRp, а CoV2 и панголин-2019 имеют несколько общих мутаций, не наблюдаемых в RaTG13:
Я также решил сравнить кодоновый профиль между RaTG13 и другими штаммами из R. affinis, живших в той же заброшенной шахте. К сожалению, для других штаммов Ra были опубликованы только 816-нуклеотидные сегменты из гена RdRp (RaBtCoV/3750 и RaBtCoV/4307–2), поэтому для сравнения кодоновых предпочтений я извлек соответствующий 816-нуклеотидный сегмент из RaTG13. В итоге, RaTG13 опять заметно отличается, в то время как два других штамма довольно близки: