DeepMind AlphaFold2 и Github Copilot являются одними из наиболее ярких успехов науки и техники в 2021 году. Спустя два года после исходного прорыва AlphaFold, специалисты из DeepMind выпустили вторую версию их революционной системы предсказания 3D-структуры белков по последовательности. На сей раз им, фактически, удалось решить задачу фолдинга белка, которая оставалась неразрешенной более 50 лет. В этом посте я постараюсь кратко описать принцип действия этой системы, как я его вижу.
Содержание
Постановка задачи и принцип действия
Поток данных в системе в целом
Последовательности: базы данных Uniprot, Mgnify; пакет программ HMMER
Структуры: база данных PDB; пакеты программ HHpred, OpenMM
Evoformer: эмбеддинги множественных выравниваний и парные представления, а также их итеративное улучшение
Структурный модуль: 3D структура из эмбеддинга последовательности
Механизм внимания и трансформерная архитектура
Механизм внимания на масштабированном скалярном произведении: Запросы (Queries), Ключи (Keys) и Значения (Values), нестрогий поиск в базе данных ключ-значения
Многоголовое внимание на векторах эмбеддингов аминокислотных остатков
Evoformer: уточнение эмбеддингов множественных выравниваний и парных представлений
Внимание по осям (Criss-cross (axial) attention)
Внимание по колонкам выравнивания, дополненное использованием информации из парных представлений
Внимание по последовательностям выравнивания
Уточнение парного представления из уточненного эмбеддинга выравнивания через внешнее произведение
Уточнение парного представления через мультипликативный модуль неравенства треугольника
Уточнение парного представления через механизм внимания, реализующий неравенство треугольника
Структурный модуль
Внимание с неподвижной точкой (Invariant Point Attention, IPA)
Уточнение остова
Релаксация структуры физическими методами (OpenMM)
Функции потерь: FAPE и дополнительные функции потерь
Переработка (refinement/recycling)
Самодистилляция (self-distillation)
Протокол обучения и исследование абляции
Постановка задачи
Белки - это линейные полимеры, состоящие из мономеров - аминокислот - и складывающиеся в пространстве в сложные трехмерные структуры. Трехмерные структуры белков позволяют понять их функцию и практически значимы для поиска/подбора новых лекарств.
Прочтение последовательности белка дешево и может быть произведено напрямую методами масс-спектрометрии (порядка $100 за образец) или секвенирования кодирующей данный белок ДНК ($100-$1000 за экзом/геном). Экспериментальное определение трехмерной структуры белка - гораздо более дорогая и трудоемкая задача ($100k-$1M за структуру), которая обычно решается методами рентгеновской кристаллографии или ЯМР.
.")
Типичный конвейер для рентгеноструктурного анализа состоит из кристаллизации белка (что не всегда возможно; так, плохо растворимые белки плохо кристаллизуются), эксперимента по рентгеновской дифракции на полученном кристалле белка, решения фазовой проблемы и создания модели белка по полученной карте электронной плотности.

Результат такого эксперимента нельзя считать абсолютной истиной, так как многие шаги в нем являются в какой-то мере искусством. Для получения структуры высокого разрешения требуется очень качественный кристалл (который может и не свариться, особенно для плохо растворимых мембранных белков). Различных методов решения фазовой проблемы немало. По одной и той же карте электронной плотности (особенно, низкого разрешения) два разных интерпретатора могут предложить структуры, существенно (процентов на 10-20%) различающиеся между собой. Наконец, белки в растворе живут и "дышат", и их рентгенные структуры похожи на "живой" белок примерно в той же мере, в какой фотографии замороженных трупов, сложенных штабелями в морге, похожи на живых людей. Уровень ошибки предсказания, достигнутый AlphaFold2 по сравнению с экспериментом, мало отличается от уровня разногласия между результатами двух разных групп, делающих расшифровку структуры одного и того же белка, поэтому я считаю, что AF2 скорее решил задачу фолдинга белка, чем не решил (пусть и с некоторыми оговорками).
Итак, от AlphaFold2 требуется предсказать 3D-структуру нового белка, имея только его последовательность. В качестве обучающей выборки можно использовать данные последовательностей и белков из общедоступных биоинформатических баз данных.
Белковые последовательности известных белков содержатся в банке данных Uniprot (где их более 200 миллионов) и базе данных MGnify (о ней - позже).
Структуры белков доступны в базе данных PDB (содержащей менее 200 тысяч структур). В ней находятся файлы .pdb со структурами белков, каждый файл содержит координаты всех атомов или только тяжелых атомов (то есть не атомов водорода) белка.
Принцип действия AlphaFold2
Методы предсказания трехмерной структуры белка in silico, основанные на чистой физике, в общем случае не способны достигать результатов, сопоставимых с экспериментом. Статистический подход оказался более действенным.
Как правило, белки консервативны в эволюции. К примеру, у человека, лошади или рыбы имеются собственные версии гемоглобина, которые исходно происходят от одного белка. Несколько различающиеся версии одного и того же белка у разных видов называются гомологами.
Основная масса событий в эволюция носят нейтральный характер (т.е. большинство мутаций не влияют на функцию белка, поэтому в последовательностях белков со временем происходит множество замен). При этом структура белков значительно консервативнее последовательности. Типичным является случай, когда последовательность белка между не очень близкими видами измениалсь на 70%, а 3D-структура белка более-менее не изменилась. Сопоставление гомологов из разных видов позволяет получить важную дополнительную информацию о белках, и обычно делается посредством записи последовательностей белков в двумерную табличку, которая называется выравниванием (multiple sequence alignment, MSA). Последовательности белков из разных видов записываются в ряды, так что соответствующие друг другу аминокислотные остатки находятся в одних колонках.
 и соответствующая 3D-структура белка (справа). На выравнивании отмечены вариабельные, консервативные и коэволюционирующие позиции. Информация о коэволюции может предполагать взаимодействие между коэволюционирующими аминокислотными остатками и то, что они сближены в пространстве.")
Консервативность позиции в выравнивании обычно означает, что такая позиция важна для функции белка, например, для сохранения структуры, каталитической активности или узнавания сайтов связывания. Грубо говоря, если в данной позиции выравнивния не бывает замен, то все организмы, в белках которых случилась замена в данной позиции, вымерли, а значит, эта позиция не может меняться по какой-то веской причине.
Коэволюция двух аминокислотных остатков часто предполагает взаимодействие между ними (и, вероятно, сближенность их в пространстве). Эта информация может служить основой для предсказания 3D-структуры белка.
Хочу также коротко отметить роль метагеномики в недавних успехах предсказания 3D-структур. Обычно белки в базах данных вроде Uniprot происходят из организмов, которые удается культивировать в лабораторных условиях и секвенировать. Однако, очень большое количество микроорганизмов в лабораторных условиях выращивать не удается, а разнообразие белков в них огромно. Поэтому в 2000-ые годы некоторые исследователи пошли по другому пути: они стали секвенировать не отдельные виды, а просто зачерпывать образцы из среды, и секвенировать всю ДНК из этого бульона. Оказалось, что такие "метагеномные" образцы содержат массу новых гомологов известных белков, и использование данных коэволюции из них повысило качество предсказания 3D-структур белков в середине 2010-ых. AlphaFold2 использует такую метагеномную информацию из базы данных MGnify, которая используется как дополнение к Uniprot.
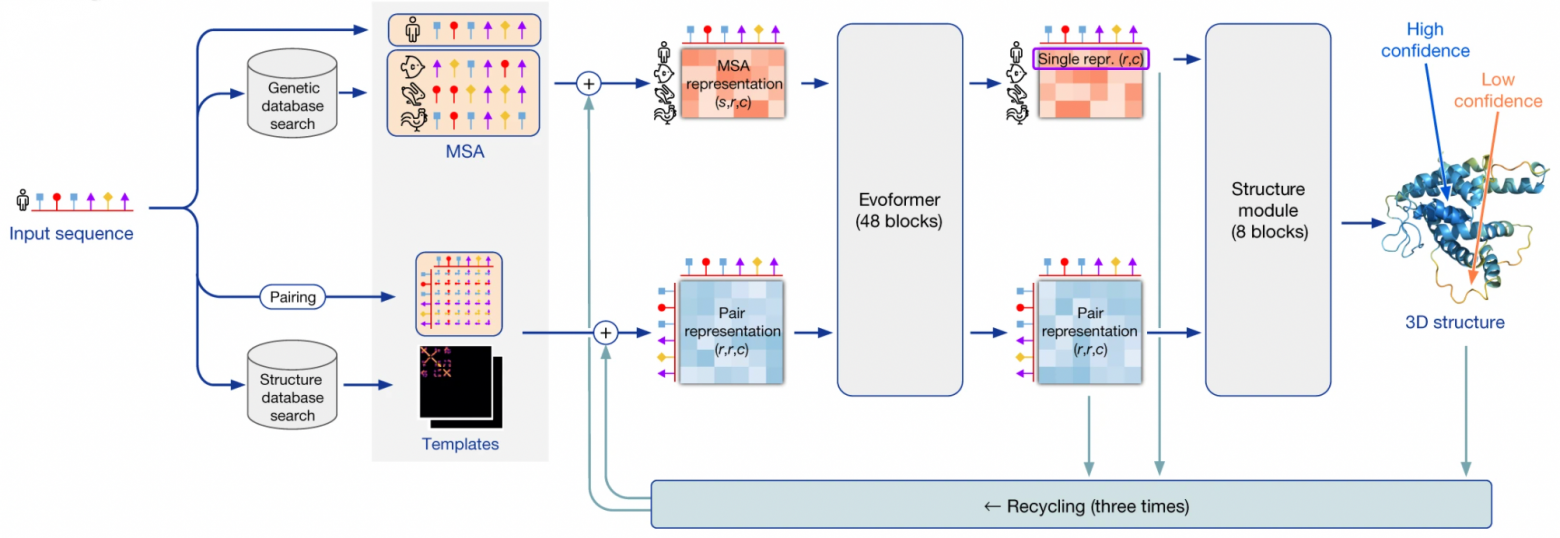
Конвейер AlphaFold2
Я использую термин “AlphaFold2” в двух контекстах: в широком смысле это вся система предсказания трехмерной структуры, использующая множество сторонних программ и баз данных. В узком смысле это нейронная сеть, лежащая в сердце этого конвейера.

Система AlphaFold2 получает на вход последовательность белка. На первом шаге она использует программы из пакета HMMER для поиска гомологов входной последовательности в базах данных Uniprot и MGnify. Пакет HMMER был создан в начале 2000-ых, и основан на скрытых Марковских моделях/Марковских цепях. Тогда этот подход доминировал в распознавании/синтезе речи, и биоинформатики позаимствовали его для своих нужд. HMMER находит гомологи данного белка в базах и возвращает выравнивание всех гомологов, которые он нашел.
AF2 также использует пакет HH-suite для поиска 3D-структур по банку данных PDB, которые могли бы соответствовать гомологам поданной на вход последовательности. Если нам повезло, и для какого-то из гомологов последовательность белка уже экспериментально расшифрована, задача кратно упрощается - фактически, остается только провести гомологичное моделирование, то есть, грубо говоря, натянуть последовательность нашего белка на эту структуру. Однако, менее чем для 0.1% белков имеется расшифрованная структура, поэтому в общем случае нужно ожидать, что такой шаблон структуры будет недоступен.
Если 3D-структура все-таки нашлась, то на основе ее AF2 строит так называемую таблицу парного представления остатков - квадратную таблицу, где каждый элемент описывает взаимодействие i-ой аминокислоты с j-ой. Если 3D-структуры не нашлось, то такая таблица инициализируется разумными значениями по умолчанию.
Далее AF2 генерирует векторные эмбеддинги из аминокислотных остатков в выравнивании и из пар остатков в парном представлении. Я не буду углубляться в то, как это делается, но нетрудно придумать несколько способов. Только замечу, что для тонких выравниваний, в которых менее 30 последовательностей, AlphaFold2 работает не очень хорошо - недостаточно данных. Если же число последовательностей в выравнивании сильно больше сотен, то это тоже плохо, потому что сложность алгоритма растет квадратично от размера входа. В таком случае делается кластеризация наиболее близких последовательностей.
Далее следует ядро AlphaFold2: end-to-end нейронная сеть, основанная на архитектуре трансформер. Нейросеть получает на вход эмбеддинги выравнивания и парного представления, итеративно улучшает их на все более глубоких слоях, и возвращает 3D-структуру, предсказанную на их основе.
Нейросеть состоит из 2 ключевых суб-модулей:
Модуль Evoformer состоит из 48 однотипных блоков и итеративно улучшает эмбеддинги выравнивания и парного представления, по сути, детектируя все более абстрактные паттерны взаимодействия между аминокислотными остатками .
Структурный модуль нейросети, состоящий из 8 однотипных блоков, предсказывает 3D-структуру белка на основе эмбеддинга нашей последовательности (он вырезается из эмбеддинга полного выравнивания), и эмбеддинга парного представления.
После того, как некая 3D-стукрутра была получена на выходе из нейросети, дополнительно используется пакет OpenMM, который на основании физических методов релаксирует полученную структуру.
Данная процедура повторяется трижды в ходе процесса под называнием "переработка" (recycling).
Механизм внимания и трансформерная архитектура
Как было сказано выше, нейросеть, лежащая в основе AlphaFold2, основана на трансформерной архитектуре и механизме внимания. Я коротко объясню их далее. Смело пропускайте этот раздел, если уже знакомы с этими конструкциями.
Механизм внимания получил широкую популярность в районе 2014-2015 в задачах машинного перевода (к примеру, с английского на французский). На тот момент лучшие архитектуры были основаны на рекуррентных нейросетях (RNN).
Сначала обнаружилось, что добавление к ним механизма внимания улучшает качество перевода. Позднее выяснилось, что от рекуррентной нейросети можно избавиться вовсе, и наслоив несколько однотипных блоков, основанных на механизме внимания, можно добиться примерно тех же результатов, что и с рекуррентными нейросетями, используя в 100 раз меньше вычислительных ресурсов.
К 2017 сформировалось понимание, что внимание - это базовый строительный блок нейросетей, альтернативный сверточным и рекуррентным нейросетям, который может успешно использоваться в обработке сигналов самых разных модальностей, в том числе изображений, текстов, звука и т.д.
Архитектуры, основанные только на механизме внимания, называются трансформерами. Блок Evoformer AlphaFold2, как подсказывает его название, является частным случаем трансформера (да и структурный модуль, на самом деле, тоже).
Механизм внимания
Механизм внимания формулируется в терминах нестрогого поиска в базе данных ключ-значение.
Предположим, что у нас есть база данных ключ-значение (к примеру, просто питоновский словарь), и мы делаем запрос к ней, в котором содержится орфографическая ошибка.
Мы хотим, чтобы наша база данных сравнила запрос с каждым ключом базы данных, и вернула взвешенную сумму значений, такую что вес каждого значения равен вероятности того, что пользователь имел в виду соответствующий ему ключ.

# Suppose that we have a key-value database and implement a fuzzy search in it database = { "Ivanov": 25, "Petrov": 100, "Sidorov": 5 } query = "Pvanov" # note the typo in query def hamming_distance(query: str, key: str) -> int: distance = 0 for i, _ in enumerate(query): if query[i] != key[i]: distance += 1 return distance # construct a vector of similarities between query and keys # similarity=1 if query=key; # similarity=0 if there is nothing in common between query and key similarities = {} for key in database: similarities[key] = (1 - hamming_distance(query, key) / len(query)) print(f"Similarities before softmax: {similarities}") output = 0 for key, value in database.items(): output += similarities[key] * value output
Similarities before softmax: {'Ivanov': 0.8333333333333334, 'Petrov': 0.5, 'Sidorov': 0.0} 70.83333333333334
В данном примере кода содержится одна неточность: вместо вероятности, что под запросом пользователь понимал ключ, мы измеряли просто сходство между запросом и ключом. Таким образом сумма этих мер сходства больше 1, и их нельзя интерпретировать как вероятности.
Поэтому требуется нормализовать эту меру сходства, так, чтобы сумма равнялась 1. Для этого используется функция Softmax:

где Softmax задается как:

# However, note that the sum of similarities in previous solution was more than 1. # We probably want to preserve the sum of similarities equal to 1. # Hence, apply Softmax to similarities vector to normalize it. import math def softmax(weights: dict) -> dict: """Transform a dict of weights into a dict with sum of weights normalized to 1.""" # Calculate statistical sum (softmax denominator) zustandssumme = 0 for key, weight in weights.items(): zustandssumme += math.exp(weight) return { key: math.exp(weight)/zustandssumme for key, weight in weights.items() } similarities = softmax(similarities) print(f"Similarities after softmax: {similarities}") output = 0 for key, value in database.items(): output += similarities[key] * value output
Similarities after softmax: {'Ivanov': 0.4648720549505913, 'Petrov': 0.33309538278287776, 'Sidorov': 0.202032562266531} 45.94150246338521
Softmax - это по сути мультиномиальный аналог сигмоиды. Подобно тому, как функция сигмоиды используется в логистической регрессии чтобы конвертировать значения из произвольного диапазона в диапазон [0,1], так что их можно интерпретировать как вероятности, Softmax делает то же самое в мультиномиальном случае. Другой способ думать о Softmax - это рассмотреть его как обобщение распределения Больцмана из статфизики.
Здесь следует сделать два замечания.
Во-первых, вместо одиночного запроса, как в приведенном выше примере, механизм внимания обычно принимает на вход целый набор (вектор) запросов и возвращает по значению-взвешенной сумме для каждого из них.
Во-вторых, обычно запросы, ключи и значения - это векторы. В качестве меры сходства между векторами можно использовать разные функции, например, косинусное расстояние, но обычно в механизмах внимания используется скалярное произведение (обычно поделенное на нормировочный множитель).
Вот изображение механизма внимания из классической статьи “Attention is all you need paper” (игнорируйте блок "Mask" на этой картинке, он не актуален в данной задаче):
. На вход принимаются матрицы запросов (Q), ключей (K) и значений (V), каждым столбцом которых является один вектор запроса, вектор ключа и вектор значения. Далее блок MatMul вычисляет скалярные произведения между каждой парой (запрос, ключ). Блок Scale нормирует каждое скалярное произведение из технических соображений. Блок Mask опционален и не используется в AlphaFold2. Блок Softmax применяет Softmax к каждому вектору нормированных скалярных произведений, соответствующих одному запросу и разным ключам, переводя их в такое пространство, что их сумма равна 1. Блок MatMul для каждого вектора-запроса в Q возвращает вектор-взвешенную сумму векторов значений (V) с коэффициентами, равными выходу блока Softmax для данного вектора-запроса.")
Например, вход механизма внимания может состоять из списка из 3 запросов, 3 ключей и 3 значений, где каждый индивидуальный запрос, ключ и значение представляют собой вектор-эмбеддинг размерности 256.



Вот реализация механизма внимания на PyTorch:
import torch import torch.nn as nn import torch.nn.functional as F def attention(query, key, value, mask=None, dropout=None): """Compute 'Scaled Dot Product Attention'. Mostly stolen from: http://nlp.seas.harvard.edu/2018/04/03/attention.html. """ # MatMul and Scale d_k = query.size(-1) scores = torch.matmul(query, key.transpose(-2, -1)) / math.sqrt(d_k) # Mask (optional) if mask is not None: scores = scores.masked_fill(mask == 0, -1e9) # Softmax p_attn = F.softmax(scores, dim = -1) # MatMul if dropout is not None: p_attn = dropout(p_attn) return torch.matmul(p_attn, value), p_attn
Внимание со множеством голов
Предположим, что ваши запросы, ключи и значения являются идентичными векторами (например, эмбеддингами аминокислот):

Предположим, что первая координата x_{i,1} отражает растворимость аминокислоты, вторая координата, x_{i,2} - ее размер, третья координата x_{i,3} - ее положительный заряд, четвертая координата x_{i,4} - отрицательный заряд.
Нам может потребоваться установить различные отношения между аминокислотами. Например, насколько взаимозаменяемы аминокислоты S и D в разных качествах?
Например, если нам нужна маленькая растворимая аминокислота, они вполне взаимозаменяемы. Если нам нужна заряженная аминокислота, они не одинаково хороши в этом качестве.
Давайте попробуем спроецировать данную аминокислоту на признаковое пространство "заряженная большая аминокислота". Наше признаковое пространство будет иметь размерность 2, где первая координата будет соответствовать абсолютной величине заряда, а вторая координата - размеру:


Теперь применим механизм внимания в разрезе этого отношения к 2 аминокислотам. Сходство между ними будет записываться как:

Таким образом, если мы хотим найти несколько разных отношений сходства между аминокислотами, мы просто будем использовать несколько разных матриц проекции на разные признаковые пространства.
Матрица проекции + следующий за ней механизм внимания вместе носят название головы внимания. Нейросеть будет обучаться детектировать разные отношения между эмбеддингами аминокислот с помощью нескольких голов внимания на каждом слое Эвоформера. Обычно на каждом слое используется h голов.
В качестве запросов, ключей и значений описанного выше механизма внимания мы будем подавать один и тот же вектор эмбеддинга X_i. Такой механизма внимания, где все три сущности Q, K, V = X_i совпадают, называется самовнимание (self-attention). Все эти входы каждая голова будет проецировать на ее признаковое пространство матрицей W, а потом будет применять описанный выше механизма внимания.
, ключей (K) и значений (V). В случае самовнимания все они - одна и та же матрица X. К ним применяется h проекций на признаковые пространства h голов с помощью матриц линейного преобразования W_h (блок Linear), далее каждая голова применяет механизм внимания (блок Scaled Dot-Product Attention), результаты работы каждой головы конкатенируются в единый массив (блок Concat), и он проецируется на исходное пространство еще одним блоком Linear.")
После применения механизма внимания, выходы со всех голов конкатенируются в единый вектор (см. блок “Concat” на картинке).
Затем конкатенированный вектор этих выходов проецируются обратно на пространство размерности исходного эмбеддинга (блок “Linear” на картинке).
Получившийся механизм называется блоком многоголового внимания (multi-head attention). Подобные блоки являются базовым элементом нейросети-трансформера. Чем больше подобных блоков мы наслоим, тем более высокоуровневые отношения сможет идентифицировать наша нейросеть. Код на PyTorch:
import torch import torch.nn as nn import torch.nn.functional as F class MultiHeadedAttention(nn.Module): """Mostly stolen from: http://nlp.seas.harvard.edu/2018/04/03/attention.html.""" def __init__(self, h, d_model, dropout=0.1): "Take in model size and number of heads." super(MultiHeadedAttention, self).__init__() assert d_model % h == 0 # We assume d_v always equals d_k self.d_k = d_model // h self.h = h self.linears = clones(nn.Linear(d_model, d_model), 4) self.attn = None self.dropout = nn.Dropout(p=dropout) def forward(self, query, key, value, mask=None): """Implements multihead attention""" if mask is not None: # Same mask applied to all h heads. mask = mask.unsqueeze(1) nbatches = query.size(0) # 1) Do all the linear projections in batch from d_model => h x d_k query, key, value = \ [l(x).view(nbatches, -1, self.h, self.d_k).transpose(1, 2) for l, x in zip(self.linears, (query, key, value))] # 2) Apply attention on all the projected vectors in batch. x, self.attn = attention(query, key, value, mask=mask, dropout=self.dropout) # 3) "Concat" using a view and apply a final linear. x = x.transpose(1, 2).contiguous().view(nbatches, -1, self.h * self.d_k) return self.linears[-1](x)
Здесь я опускаю некоторые важные технические инженерные моменты, такие как применение LayerNorm и Residual connections между блоками multi-head attention, нормировку Scaled Dot-Product Attention, Positional Encoding и т.д.
Модуль Evoformer
Модуль Evoformer состоит из 48 идентичных блоков, которые принимают эмбеддинги множественного выравнивания и парного представления на вход и возвращают их усовершенствованную версию на выходе.
 общая архитектура; (b) матрица/тензор эмбеддинга парного представления; (c) неравенства треугольника.")
Эвоформер состоит из 3 основных шагов:
Эвоформер улучшает эмбеддинг выравнивания вниманием по осям (axial/criss-cross), используя дополнительную информацию, содержащуюся в парном представлении.
Эвоформер улучшает парное представление с помощью обновленного на предыдущем шаге эмбеддинга выравнивания, используя блок среднего внешних произведений.
Эвоформер применяет блоки неравенств треугольника к парному представлению, обновленному на предыдущем шаге, чтобы обеспечить согласованность данных.
Разберем каждую из частей эвоформера по отдельности.
Evoformer: улучшение множественного выравнивания

МЕХАНИЗМ ВНИМАНИЯ ПО ОСЯМ В ЭВОФОРМЕРЕ
MSA update part of Evoformer uses an approach, called axial or criss-cross attention. It was suggested in visual transformers circa 2020. In visual transformers attention needs to be applied to each pixel of an image, and using the full 2D image as keys would be computationally inefficient.
A frugal alternative to full 2D attention, is to first attend to all pixels in the same row as the query pixel and then attends all pixels in the same column. Same approach was employed here by evoformer. It first attends to other aminoacid residues in the same sequence (which is called row-wise gated self-attention), and then residues from other sequences in the same column (column-wise gated self-attention).
САМОВНИМАНИЕ С ЗАТВОРОМ ПО ПОЗИЦИЯМ ВЫРАВНИВАНИЯ
 выравнивания в Эвоформере")
Считаю нужным обсудить 2 аспекта самовнимания по колонкам выравнивания с механизмом запирания/затвора.
Во-первых, обратите внимание на то, что к матрице внимания прибавляется проекция матрицы парного представления. Это логично: если по мере формирования трехмерной структуры выясняется, что два остатка сближены и взаимодействуют, это будет отражено в матрице парного представления, и, в свою очередь, повлияет на матрицу внимания, увеличив вес взаимодействия соответствующих остатков в ней.
Во-вторых, верхняя стрелка на этой схеме соответствует механизму запирания на этой схеме, о котором я еще не говорил. Механизм запирания похож на затвор транзистора - если в векторе затвора "закрыт" некий элемент, это практически обнуляет соответствующий элемент выходного вектора механизма внимания. Затвор будет закрыт, если проекция эмбеддинга соответствующей аминокислоты имеет малую “длину” (длину в кавычках, потому что "вектор" затвора все-таки имеет c каналов, и запирание происходит для каждого канала в отдельности). Технически для запирания используется все та же сигмоидная функция от проекции эмбеддинга позиции выравнивания, которая будет близка к 1, если значение координаты проекции достаточно велико или близка 0, если оно достаточно мало. Соответственно, в первом случае затвор будет открыт, а во втором - открыт. Однако, назначение этого механизма для меня неочевидно, и может быть просто инженерным приемом, для которого известно, что так сеть лучше работает (см. ссылки на литературу в конце статьи).
САМОВНИМАНИЕ С ЗАТВОРОМ ПО ПОСЛЕДОВАТЕЛЬНОСТЯМ ВЫРАВНИВАНИЯ

Самовнимание по последовательностям выравнивания обеспечивает обмен информацией между последовательностями выравнивания внутри каждой колонки выравнивания. Этот шаг помогает AlphaFold2 обнаружить, что между последовательностями в данной позиции есть коэволюция, или что позиция консервативна. Кроме того, поскольку именно для первой последовательности выравнивания делается предсказание 3D-структуры белка, через нее обновленная информация о 3D-структуре распространяется на другие последовательности.
В дополнительных материалах к статьей DeepMind приводит визуализацию карт внимания на этом блоке (см. изображение ниже). Ряд (a) на картинке показывает, что для каждой позиции выравнивания имеется какое-то подмножество последовательностей, которое несет основную информацию. Ряд (b) показывает, что после нескольких итераций переработки все последовательности обращают внимание на первую последовательность (для которой предсказывается 3D-структура). Ряд (c) показывает, что промежуточные слои AF2 постепенно выучивают, по сути, филогенетическое дерево, описывающее эволюционное родство последовательностей (см. правую нижнюю картинку “Hamming distance”, треугольную матрицу над главной диагональю которой можно рассматривать как филогенетическое дерево).
-(c) см. в тексте статьи. Картинка (d) содержит предсказанные расстояния между парами C-альфа атомов аминокислотных остатков белка. Картинка (e) содержит матрицу расстояний Хэмминга между последовательностями выравнивания, фактически, эквивалентную филогенетическому дереву, описыающему эволюционное родство данных последовательностей.")
Приведу интерпретацию этих карт внимания из дополнительных материалов к статье:
In Suppl. Fig. 13 we show a visualization of the attention pattern in the MSA along the columns a h_{sti}hsti (line 4 of Algorithm 8). We slice along the last axis ii and display the stst array as heat map.
The original MSA subset shown to the main part of the model is randomly sampled. As such the order is random except for the first row. The first row is special because it contains the target sequence and is recycled in consecutive iterations of the model. Due to the shallow MSA of this protein, the random subset leads to a random permutation of the sequences. In order to facilitate easier interpretation of the attention patterns here we reorder the attention tensor by using a more suitable order for the MSA. We perform a hierarchical clustering using the Ward method with simple Hamming distance as a metric and use the output to re-index the sequence dimension in the MSA attention tensor. We resort the indices from the hierarchical clustering manually to keep the target sequence in the first row. This manual sorting is done in such a way as to keep the tree structure valid. The Hamming distances between the reordered sequences (see Suppl. Fig. 13e) show a block-like structure quite clearly after the reordering.
The attention pattern in the first layer of the network in the first recycling iteration (e.g. layer 0, head 7 in Suppl. Fig. 13a) is not very informative and is largely averaging as can be seen by looking at the range of the attention weights.
In the same head, but in the last recycling iteration (Suppl. Fig. 13b) we see that all sequences at all positions attend to the first row. Therefore this head behaves differently upon recycling and is presumably important for distribution the information in the recycled first row to the rest of the MSA representation. Layer 6, head 6 (Suppl. Fig. 13c) shows a pattern that is fairly common in the column-wise MSA attention, here the pattern only varies lightly as one goes along the sequence and there is a clear structure in blocks of sequences that attend to each other. We note that these seem somewhat similar to the blocks derived from hierarchical clustering using Hamming distance. Whether attention patterns provide a good explanation for the behaviour of a given model or are predictive of interventions into the model is a topic of debate in the community, see [127], [128], [129]. A detailed analysis of the generality and predictivity of these attention patterns is beyond the scope of this paper.
ПЕРЕХОДНЫЙ БЛОК ВЫРАВНИВАНИЯ
Последний элемент подблока уточнения эмбеддинга выравнивания в Эвоформере - это переходный блок. Я не вполне понимаю его назначение, и оно не обсуждается подробно в статье и дополнительных материалах. Полагаю, его функция - чисто инженерная.

Эвоформер: уточнение парного представления после улучшения эмбеддинга выравнивания
После того как эмбеддинг выравнивания был улучшен в первой части Эвоформера, это улучшение следует отразить и в парном представлении.
Кажется, что самым интуитивным способом это сделать было бы просто спроецировать карты внимания из блока внимания по колонкам выравнивания на парное представление.
Однако, надо помнить, что мы должны взять среднее по всем последовательностям в данной колонке. Поэтому делается что-то подобное, просто математическим представлением этой операции является среднее по последовательностям внешних произведений эмбеддингов колонок. Напоминаю, что операция внешнего произведения (outer product) в линейной алгебре - это частный случай Кронекеровского произведения или тензорного произведения.

Эвоформер: неравенства треугольника как естественные условия для парных расстояний

Осталось обсудить последнюю часть Эвоформера - соблюдение неравенств треугольника в парных представлениях после обновления эмбеддинга парного представления из эмбеддинга множественного выравнивания.
Сначала сформулируем, чего мы хотим достичь с помощью неравенств треугольника? Для каждого ребра {i,j}, мы хотим рассмотреть все пары ребер {i,k} and {k,j} и убедиться, что для всех k длина d{i,j} ребра {i,j} не превышает суммы длин ребер d{i,k} и d{k,j}:
d{i,j} < max(d{i,k} + d{k,j})
Однако, реализовать данную логику может быть нетривиально так, чтобы она была дифференцируема. Вместо этого в AlphaFold2 используются эмпирические версии этого ограничения, несколько более мягкие.
В AlphaFold2 есть 2 разных вида блоков, обеспечивающих неравенство треугольника: triangle multiplicative update и triangle self-attention. Исходно triangle multiplicative updates был разработан как вычислительно экономная альтернатива блокам triangle self-attention. Можно убрать любой из этих типов блоков, и система продолжит работать достаточно хорошо. Однако, оказалось, что SOTA достигается, если вставить оба вида модулей, поэтому так и было сделано.

Блок мультипликативного неравенства треугольника работает так: он берет два ряда из парного представления, i-ый и j-ый. Для каждого индекса остатка k в обоих рядах, мы проецируем эмбеддинги ребер {i,k} и {j,k} матрицей проекции на пространство более никзой размерности, применяем к ним механизм запирания и берем Адамарово произведение получившихся i-ого и j-ого векторов. Затем мы суммируем по k результирующий вектор, получая скалярную сумму, и используем ее в качестве нового значения элемента {i,j} матрицы парного представления, предварительно еще раз применив к ней нормализацию через LayerNorm и механизм запирания с исходным значением элемента {i,j} как управляющего значения затвора.
Я не могу с уверенностью утверждать, что понимаю, как работает этот модуль, но вот мои соображения. Мы видим, что вместо того, чтобы отыскать такой элемент k, для которого достигается минимум d{i,k} + d{j,k}, мы рассчитываем прореженную механизмом запирания сумму по всем остаткам:

Вероятно, эта сумма (нормализованная layer norm и прореженная механизмом запирания) дает какое-то взвешенное среднее ограничений, создаваемых парами ребер ({i,k}, {j,k}), для тех k, которые ограничивают длину ребра {i,j} сильнее всего. В итоге мы получаем какую-то аппроксимацию истинного ограничения.
Второй механизм с вниманием к неравенству треугольника видится еще мне менее интуитивным. В нем ребро, длину которого мы хотим ограничить, используется как query в механизме внимания; длины одного из ребер, например, {i,k}, используются как keys и values, в механизме внимания, а длина другого ребра {j,k} прибавляется к affinity matrix.

Очень интересно проанализировать карты внимания для механизма внимания к неравенству треугольника (см. изображение ниже).
В ряду (a) мы видим что-то похожее на паттерн свертки радиусом 4 (диаметром 8) аминокислотных остатков. К примеру, расстояние между остатками 14 и 15 зависит от расстояний между остатками 14-16, 14-17, 14-13, 14-12 и 14-11. Авторы статьи рассуждают, что данная голова внимания научилась идентифицировать водородные связи в белках, за счет которых те образуют альфа-спирали. Шаг альфа-спирали составляет как раз 4 остатка, так что остатки i-ый и (i+4)-ый взаимодействуют между собой водородными связами, и что-то похожее мы и видим на картах внимания этой головы.
В ряду (b) мы видим, что для 14-ой аминокислоты все расстояния от нее до других аминокислот - 14-1, 14-2, …, 14-100 - зависят от длины связей 14-28 и 14-80. Автооры объясняют, что остатки 14, 28 и 80 - это все цистеины, которые способны образовывать дисульфидные SS-мостики. Таким образом, наличие или отсутствие дисульфидного мостика между остатками 14-28 или 14-80 во-многом влияет на расстояния между 14-ой аминокислотой и всеми остальными аминокислотными остатками в данном белке, и этот паттерн и выучилась распознавать данная голова.
Наконец, ряд (c) на изображении указывает на то, что достаточно рано, уже к 11-ому слою Эвоформера, в нейросети начинает формироваться довольно неплохое представление об общей трехмерной структуре белка и парном представлении. Сравните предсказанную матрицу парного представления расстояний между C-альфа атомами (самая правая картинка в первом ряду, Predicted CA-CA distances) с нижним рядом (c). Это предположение хорошо подтверждается исследованием абляции, где структурный модуль шунтом присоединяют к промежуточным слоям Эвоформера, и показывают, что для белков с достаточно простой 3D-структурой уже к 10-20-ому слоям очень хороший эскиз 3D-структуры успевает сформироваться (см. картинку исследование абляции в самом конце статьи, пункт b).
-(c) см. в тексте статьи. Ряд (d) содержит предсказанные расстояния между парами C-альфа атомов аминокислотных остатков белка.")
Опять же, привожу цитату из дополнительных материалов статьи:
In this section we are going to analyse a small subset of the attention patterns we see in the main part of the model. This will be restricted to relatively short proteins with fairly shallow MSA’s in order for easier visualization.
We are going to examine the attention weights in the attention on the pair representation in the “Triangular gated self-attention around starting node” (Algorithm 13). The attention pattern for a given head h is a third order tensor a h_{ijk}hijk (line 5 of Algorithm 13), here we will investigate different slices along the i axis as well as averages along this axis, and display the jk array as a heat map. We specify the attention pattern for a specific head by the recycling index r, layer index l and head index h. We show the attention patterns in Suppl. Fig. 12
In layer 0, head 0 (Suppl. Fig. 12a) we see a pattern that is similar to a convolution, i.e. the pair representation at (i, i + j) attends to the pair representation (i, i + k), where j and k are relatively small and the pattern is fairly independent of i. The radius of the pattern is 4 residues, we note that the hydrogen bonded residues in an alpha helix are exactly 4 residues apart. Patterns like this are expected in early layers due to the relative position encoding, however one would naively expect it to be wider, as the relative positions run to 32.
In layer 2, head 2 (Suppl. Fig. 12b) we see a very specific attention pattern. We see that positions 14, 28 and 80 play a special role. These correspond to the positions of cysteine residues in the protein. We see that at the cysteine positions the attention is localized to the other cysteine positions as exemplified by the bands in the first two panels. Meanwhile away at positions different from the cysteine like position 20, we see that the attention is devoid of features suggesting that the model does not use the attention at these positions. We found this behaviour to be consistent across different positions and proteins, this possibly indicates finding possible disulfides as a key feature of some heads.
A head later in the network (Suppl. Fig. 12c) shows a rich, non-local attention pattern resembling the Suppl. Material for Jumper et al. (2021): Highly accurate protein structure prediction with AlphaFold 55 distance between pairs in the structure (Suppl. Fig. 12d).
Структурный модуль
Мы закончили с обсуждением того, как в Эвоформере происходит итеративное улучшение эмбеддингов выравнивания и парного представления, и теперь пора перейти непосредственно к тому, как делается предсказание 3D-структуры.
На вход структурный модуль берет эмбеддинг первой последовательности, вырезанной из выравнивания, для которой делается предсказание 3D-структуры белка, и парное представление.
Центральной частью этого процесса является модуль внимания неподвижной точки (Invariant point attention, IPA), в котором происходит обмен информации между всеми тремя форматами. Однако интересно, что если вовсе убрать этот элемент, но сохранить процесс переработки, AlphaFold2 продолжает работать на удивление хорошо (см. исследование абляции).
 архитектура структурного модуля в целом; (e) формализация представления остова белка как набора треугольников - элементов остова; (f) представление одиночного аминокислотного остатка в остове белка парой (Матрица, вектор) - (R,t) (Rotation, translation) - (Поворот, сдвиг).")
Перед тем как перейти к описанию принципа работы структурного модуля, следует сказать несколько слов о структурной биологии белков.
Пептидная цепь белка образована аминокислотами. Каждый аминокислотный остаток состоит из 3 обязательных частей - аминогруппы, C-альфа атома и группы карбоновой кислоты. Эти 3 группы образуют так называемый остов белка, и именно трехмерную структуру остова важно корректно предсказать в первую очередь. Помимо того, у каждой аминокислоты есть радикал, который разный у разных типов аминокислот (обычно встречается 20 типов стандартных аминокислот - у которых есть 20 типов радикалов).
С учетом вышесказанного расположение каждого аминокислотного остатка в пространстве может быть описано 3 торсионными углами - φ, ψ и ω. Первые два угла - φ и ψ - обладают большой свободой вращения, в то время как угол ω не может сильно отличаться от 180 градусов в связи с тем, что у атома азота аминогруппы имеется электронная пара, которая любит лежать в одной плоскости с пи-электронным облаком двойной связи C=O карбоксильной группы. Поэтому пептидная связь N-C ведет себя почти как двойная и вращение вокруг нее затруднено.
Что касается радикалов аминокислот, их длины рознятся, поэтому вращение вокруг связей между атомами боковой цепи радикалов описывается для каждой аминокислоты переменным числом торсионных углов χ1, χ2, …, вплоть до χ5.
. Отмечены торсионные углы φ, ψ и ω. Радикалы аминокислот обозначены R1 и R2 и связаны с С-альфа атомами.")
Исходя из этого DeepMind решили представить каждый аминокислотный остаток треугольником, который они называют элемент остова (backbone frame), где вершина треугольника при тупом угле соответствует C-alpha атому, а две других вершины треугольника соответствует атому азота аминогруппы и атому углерода карбоксильной группы. Для каждого элемента остова требуется предсказать 3-вектор сдвига (насколько нужно сдвинуть этот треугольник относительно начала координат) и матрицу поворота 3x3 (угол поворота треугольника). Основная нейросеть AF2 предсказывает только расположение остова, в то время как за углы χ, описывающие радикалы, отвечает отдельная нейросеть ResNet.
В начальный момент работы системы все элементы остова помещаются в начало координат (DeepMind придумали для этого метода забавное и красноречивое название “инициализация черной дырой”), и далее их координаты обновляются структурным модулем на основании информации, полученной от модуля внимания неподвижной точки.
Механизм внимания неподвижной точки отвечает за агрегацию информации из 3 источников - эмбеддинга последовательности (вырезанного из эмбеддинга выравнивания), парного представления и элементов остова. Эта объединенная информация обогащает эмбеддинг последовательности, из которого после модуля IPA делается обновленное предсказание структуры. В матрицах внимания все три источника информации суммируются, и затем вносят вклад в обновление эмбеддинга последовательности.
. Объединяем 3 источника данных: эмбеддинг последовательности, вырезанный из эмбеддинга выравнивания, парное представление и 3D-структуру остова белка и используем их для построения матрицы внимания, с помощью которых вычисляем веса значений, которыми уточняем эмбеддинг последовательности.")
Сам механизм внимания неподвижной точки не обновляет сдвиг/поворот аминоксилотных остатков. За это отвечает отдельный подблок уточнения остова в структурном модуле, который следует за механизмом внимания неподвижной точки и техническим переходным блоком. Подблок уточнения остова устроен довольно просто: сдвиг локальной системы координат каждого остатка в нем описывается просто как линейная проекция эмбеддинга данного остатка. Поворот локальной системы координат данного остатка тоже получается из эмбеддинга данного остатка, но немного сложнее (технически, все вычилсения делаются не в терминах матриц поворота, а в терминах кватернионов).
После того, как нейросетевая часть закончила предсказание 3D-структуры, запускается софт из пакета OpenMM, который далее релаксирует полученную структуру физическими методами (думаю, это хороший подход, потому что методы молекулярной динамики работают не очень хорошо, когда стартуют находясь вдалеке от локального минимума, но когда структура уже близка к локальному минимуму, справляются гораздо лучше).
Frame Aligned Point Error (FAPE) loss and auxiliary losses
Основная функция потерь, которая используется в AlphaFold2, для оценки качества предсказания структуры называется Frame Aligned Point Error (FAPE).
FAPE инвариантна к изменению глобальной системы координат (что логично, ведь от сдвига/поворота всего белка в целом структура не изменится) и представляет собой хитрую комбинацию L2-нормы по координатам тяжелых атомов в каждом остатке и L1-нормы между разными остатками с разнообразными инженерными ухищрениями, которые, очевидно, делают ее более устойчивой к выбросам и т.п. (к примеру, в структуре белка могут иметься петли, положение которых довольно сильно варьирует, но это мало влияет на общую структуру; применение L2-нормы в чистом виде было бы чувствительно к вариациям в их расположении; могу предположить, что использование разнообразных ухищрений в FAPE позволяет избежать излишней чувствительности функции потерь к подобным вариацям). FAPE может включать или не включать аминокислотные радикалы и торсионные углы χ в них.
Однако, помимо основной функции потерь FAPE используются еще дополнительные лоссы, из которых собирается линейная комбинация:

LFAPE - это FAPE loss
Laux - это лосс по дополнительным метрикам в структурном модуле
Ldist - это лосс, измеряющий качество предсказания дистограмм. AF2 пытается предсказывать не просто расстояния между остатками, но и распредения этих расстояний, называемые дистограммами. Ошибки в этих распределениях являются частью общей функции потерь.
Lmsa - это BERT-подобный лосс, измеряющий качество предсказания аминокислоты выравнивания, если ее маскировать. AlphaFold2 маскирует или мутирует позиции выравнивания и пытается предсказать, что там было исходно.
Lconf - это лосс, измеряющий степерь уверенности модели в метрике pLDDT
Lexp resolved - это лосс от головы, предсказывающей, пришла ли данная структура из эксперимента или псевдоразметки
Lviol - это лосс, измеряющий наличие структурных отклонений, таких как неправдоподобные длины связей или торсионные углы, стерические затруднения и т.п.
Уточнение структуры или "Переработка" (Refinement a.k.a. “recycling”)
Однако, как ни удивительно, все эти премудрости структурной биоинформатики, которыми пользуется структурный модуль, вносят даже меньший вклад в итоговый результат, чем процесс уточнения структуры или "переработки", как показывает исследование абляции. Без модуля внимания неподвижной точки, но с переработкой AlphaFold2 все еще функционирует очень прилично. Более того, если убрать "переработку", но оставить внимание неподвижной точки, результат будет хуже, чем если сделать наоборот - убрать внимание неподвижной точки и оставить "переработку"!
Идеологически подход переработки позаимствован из задачи оценки позы (см. статью Human Pose Estimation).

Чтобы точно предсказать позу человека, мы берем на вход фотографию человека и позу по умолчанию, конкатенируем их между собой как различные каналы, и позволяем нейросети инкрементально уточнять позу. Вполне логично, что задача предсказания расположения остова белка в пространстве не так сильно отличается от задачи оценки позы человека на фотографии.
У меня возникло субъективное предположение, что подобно тому, как в случае с неравенством треугольника команда DeepMind испробовала два альтернативных подхода, и в итоге оставила оба, что-то подобное могло произойти и с предсказанием расположения остова. В какой-то мере использование внимания неподвижной точки и переработки - это альтернативные подходы для предсказания расположения остова, однако практика показала, что оптимальный результат достигается, если оставить оба и сделать их взаимодополняющими.
Самодистилляция
Самодистилляция - это еще один инженерный прием, который позвоили DeepMind пробить baseline (см. исследование абляции).
Поскольку в доступном датасете (более 200 миллионов последовательностей) лишь небольшая часть данных имеет разметку (менее 200 тысяч структур), есть смысл рассматривать проблему как задачу обучения с частичным привлечением учителя (semi-supervised learning). Команда DeepMind избрала следующий подход.
Noisy Student Training
См. статью Self-training with Noisy Student improves ImageNet classification.
Подход Noisy Student Training недавно позволил установить новый state-of-the-art на ImageNet, подняв top-1 accuracy на 2% до 88.4% благодаря использованию 3.5 миллиардов неразмеченных изображений с Instagram.
Noisy Student Training это подоход с частичным привлечением учителя. Он дополняет идеи само-обучения и дистилляции использованием моделей-учеников равного или большего размера, чем учитель, и добавлением шума во время обучения ученика. Он включает 3 основных шага:
Обучаем нейросеть-учителя на размеченных данных
Используем учителя чтобы сгенерировать псевдо-разметку на неразмеченных изображениях
Обучаем модель-ученика на комбинации изображений с разметкой и псевдо-разметкой.
Алгоритм делает несколько итераций, обучая ученика и используя его как учителя, чтобы заново разметить неразмеченные изображения, на которых будет учиться новый ученик.
Noisy Student Training - это дополнительное усовершенствование итеративного самообучения и самодистилляции. Во-первых, каждый следующий ученик как минимум не меньше учителя, что позволяет ученику обучаться на увеличившемся датасете. Во-вторых, добавляется шум, затрудняющий ученику обучение на псевдоразмеченном датасете. В качестве шума используются аугментация типа RandAugment, и зашумление самой подели с помощью dropout'а и стохастической глубины модели (подход к регуляризации в ResNet'ах, когда на этапе обучения стохастически выкидывается часть слоев).
 и обучаем на этих зашумленных данных с псевдоразметкой, созданной учителем, нейросеть-ученика не меньшего размера, чем учитель. Повторяем несколько раз.")
Протокол обучения
AF2 обучалась на TPUv3 около недели, и затем донастраивалась в течение 4 дней.
Любопытно, что модель занимает 20GB видео памяти, а на TPUv3 доступно только 16GB. Пришлось выкручиваться: после прямого прохода часть активаций "забывали" и пересчитывали их на обратном проходе. Таким образом, пришлось жертвовать FLOPS чтобы уложиться в VRAM.

Исследование абляции
На картинке (а) ниже видно, какой вклад в качество работы AF2 вносят различные ее элементы. На картинке (b) видно, что будет, если присоединить структурный модуль к промежуточному слою Эвоформера для 3 белков - простого, среднего и сложного. Видно, что структура простого и среднего белков вырисовывается уже после несколких десятков слоев Эвоформера, а вот для сложного белка важно провести как можно больше итераций - слоев Эвоформера и циклов переработки.
 вклад различных модулей системы в общий результат (b) формирование эскиза 3D-структуры на промежуточных слоях Эвоформера для простого, среднего и сложного белка: видно, что для простого и среднего белков структура практически сформирована к 30-ому блоку Эвоформера, в то время как для сложного белка добавление дополнительных слоев Эвоформера и дополнительные итерации переработки приводят к значительным улучшениям.")
Источники
https://www.nature.com/articles/s41586-021-03819-2 - оригинальная статья AlphaFold2
https://static-content.springer.com/esm/art%3A10.1038%2Fs41586-021-03819-2/MediaObjects/41586_2021_3819_MOESM1_ESM.pdf - допонительные материалы к статье AlphaFold2
https://github.com/deepmind/alphafold - исходный код AlphaFold2
https://moalquraishi.wordpress.com/2021/07/25/the-alphafold2-method-paper-a-fount-of-good-ideas/ - блог пост Мо АльКраиши (Mohammed Al Quraishi) о AF2
http://cs371.stanford.edu/2018_slides/coevolution.pdf - команда из Стэнфорда о использовании данных коэволюции для предсказания 3D структур белков
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5493203/ - Сергей Овчинников об использовании метагеномики в предсказании 3D-структур
http://nlp.seas.harvard.edu/2018/04/03/attention.html - аннотированный трансформер
https://arxiv.org/abs/2002.05202 - статья “GLU Variants Improve Transformer”, где предложен механизм затвора/запирания (gating), похожий на использованный в AF2
https://arxiv.org/pdf/1912.00349.pdf - статья “Not All Attention Is Needed: Gated Attention Network for Sequence Data” - еще про затвор/запирение
https://openaccess.thecvf.com/content_ECCV_2018/papers/Pau_Rodriguez_Lopez_Attend_and_Rectify_ECCV_2018_paper.pdf - статья “Attend and Rectify: a Gated Attention Mechanism for Fine-Grained Recovery.” - еще про затвор/запирание
https://arxiv.org/pdf/1811.11721v2.pdf - статья “CCNet: Criss-Cross Attention for Semantic Segmentation” про осевое внимание (axial/criss-cross attention)
https://arxiv.org/pdf/2102.10662.pdf - статья “Medical Transformer: Gated Axial-Attention for Medical Image Segmentation” с использованием осевого внимания в медицинских трансформерах
https://www.youtube.com/watch?v=Uk7m93V14KA - хороший доклад по Medical Transformer на русском
https://arxiv.org/pdf/1802.08219.pdf - статья про SE(3)-эквивариантность “Tensor field networks: Rotation- and translation-equivariant neural networks for 3D point clouds”
https://arxiv.org/pdf/2006.10503.pdf - статья группы Макса Веллинга про SE(3)-эквиваринатность трансформеров “SE(3)-Transformers: 3D Roto-Translation Equivariant Attention Networks”
https://papers.nips.cc/paper/2019/file/f3a4ff4839c56a5f460c88cce3666a2b-Paper.pdf - статья “Generative models for graph-based protein design”
https://arxiv.org/pdf/1507.06550.pdf - статья “Human Pose Estimation with Iterative Error Feedback” с подоходом, аналогичным "переработке" в AF2
https://arxiv.org/pdf/1911.04252.pdf - статья “Self-training with Noisy Student improves ImageNet classification” с подохом, аналогичным самодистилляции в AF2
https://www.biorxiv.org/content/10.1101/2021.02.12.430858v1.full - статья про MSA Transformer от группы Питера Аббила
https://www.ddw-online.com/media/32/03.sum.the-cost-and-value-of-three-dimensional-protein-structure.pdf - статья 2003 года с оценкой финансового эффекта, затрат и вероятности успеха экспериментов по рентгеновской кристаллографии
https://blog.inten.to/hardware-for-deep-learning-part-4-asic-96a542fe6a81 - отличный текст о ASIC'ах для машинного от Григорий Сапунова из Intento
https://en.wikipedia.org/wiki/High_Bandwidth_Memory - об оперативной памяти в GPU
https://t.me/gonzo_ML/787 - недавний пост в телеграме о gMLP от Григория Сапунова, где обычные полносвязные перцептроны с механизмом запирания/затворами показывают резальтаты, сопоставимые с трансформерами
Благодарю Анвара Курмукова, Ольгу Золотареву и Александра Червова, а также участников сообщества SBERLOGA за помощь в подготовке данного поста. Обращаю внимание читателей, что данный текст является лишь моей попыткой интерпретации принципов работы системы AlphaFold2 и может содержать ошибки и неточности (хотя я и старался их избежать как мог).


{kind=link}