 Иногда хочется занять свой ноутбук чем-нибудь полезным, да и так чтобы с работой помогало. Несколько лет назад, в университете сталкивался бегло с моделирование методом молекулярной динамики в gromacs, это хоть и не совсем то, но уже можно было притянуть за уши к моей работе. Русскоязычной информации по запуску моделирования в gromacs удалось найти крайне мало, примерно столько же и в глобальном масштабе, все в основном обсуждают уж очень глубокие/тонкие моменты. В итоге решил разобраться в нем, а заодно и написать краткое руководство, по использованию gromacs.
Иногда хочется занять свой ноутбук чем-нибудь полезным, да и так чтобы с работой помогало. Несколько лет назад, в университете сталкивался бегло с моделирование методом молекулярной динамики в gromacs, это хоть и не совсем то, но уже можно было притянуть за уши к моей работе. Русскоязычной информации по запуску моделирования в gromacs удалось найти крайне мало, примерно столько же и в глобальном масштабе, все в основном обсуждают уж очень глубокие/тонкие моменты. В итоге решил разобраться в нем, а заодно и написать краткое руководство, по использованию gromacs. Преследуемая цель статьи — популяризировать Gromacs в широких кругах. Цели сделать полный охват метода и возможностей пакета нету, потому это будет краткий экскурс
Немного слов… или зачем нам все это надо
Если говорить о моделировании атомных или молекулярных структур, то практически все методы можно разделить на две группы: Квантово-механические методы и методы молекулярной динамики. Отличие этих групп принципиальное.
Квантово-механические методы основываются на использовании квантовой механики. Используемые в них расчеты ведутся либо из “честных/точных” квантовых уравнений, либо из их некоторых приближений, которые делаются для ускорения расчетов. Стоит отметить, что данные методы могут считать химические реакции т.е. превращения одних веществ в другие с достаточно высокой точностью. Под точностью следует понимать значения энергий связей в системе, разницы до и после реакции. Для лучшего представления точности метода знаю, что бывали ситуации когда посчитанное значение оказывалось точнее измеренного экспериментального. Т.е. более точные эксперименты в будущем подтверждали квантово-механические расчеты. Но данные методы имеют существенный недостаток это требование к большой вычислительной мощности(на “не супер компьютерах” системы в тысячу атомов имеют не приличные времена расчетов). Какие-то цифры по системам я привести не могу, подзабыл.
Молекулярная-динамика основывается на классической ньютоновской механике. Т.е. все взаимодействия считаются классическим образом из “простых” уравнений. В этом и слабость и сила метода. С одной стороны это дает возможности моделировать системы с миллионами атомов. С другой стороны расчеты идут только на уровне межмолекулярного взаимодействия и расчет хим реакций «не возможны», в отличии от квантовой химии, где считается «всё».
Примеры использования gromacs:
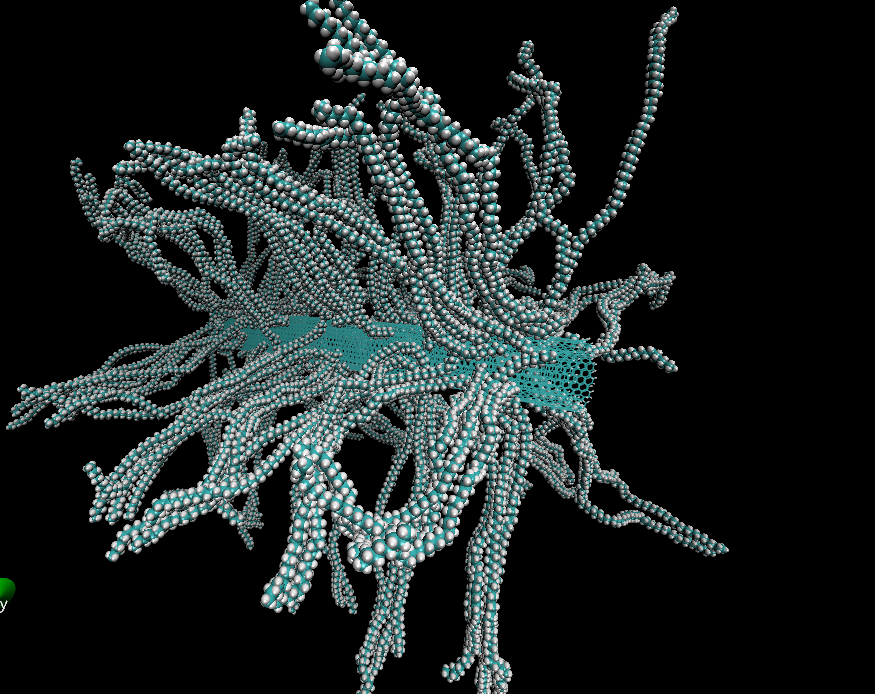
Слипание полиэтилена:
Замерзание воды
ДНК в воде
Начнем с установки, т.к. она в принципе может вызвать трудности
Установка
Установка gromacs
Можно поставить какую-то сборку из репозитория:
sudo apt-get install gromacs
но для хорошей производительности рекомендую произвести сборку самостоятельно, под свое железо(у меня это i5-3210M, geforce 620m):
почему:
при сборке указывается какие процессорные команды разрешены, использовать или нет видеокарту итп. У меня производительность собранной версии в 2 раза выше по сравнению с репозиторной, в основном конечно из-за использования видеокарты
Скачиваем последнюю, стабильную версию программы, в моем случае это gromacs 5.0.4. Распаковываем и создаем папку для сборки:
tar xfz gromacs-5.0.4.tar.gz cd gromacs-5.0.4 mkdir build cd build
далее самый важный момент, из-за которого весь этот неприглядный процесс и затеян — выбор используемых комманд процессора, методов расчета, использования видеокарты итп. Подробнее о используемых методах указано в документации к установке. По порядку, используем для настройки cmake:
- использование FFTW: -DGMX_BUILD_OWN_FFTW=ON -DGMX_FFT_LIBRARY=FFTW
- выбираем набор процессорных команд, для моего процессора это avx и соответствующая запись -GMX_SIMD=AVX_256 (другие)
- поддержка видеокарты -DGMX_GPU=ON -DCUDA_TOOLKIT_ROOT_DIR=/usr/lib/nvidia-cuda-toolkit/ путь до cuda toolkit
- -DCMAKE_INSTALL_PREFIX=/usr/local/ выбираем папку установки. Если посмотреть на значения по-умолчанию, то можно увидеть, что установка произведется в /usr/local/gromacs/, что потребует дополнительного прописывания путей, для запуска, потому просто отправляем его в /usr/local.
Настраиваем, собираем и устанавливаем:
checkinstall
при использовании chekinstall необходимо, как минимум указать название программы и её версию, чтобы не было конфликтов (gromacs 5.0.4 — у меня соответственно)
cmake .. -DGMX_BUILD_OWN_FFTW=ON -DGMX_FFT_LIBRARY=fftw3 -DGMX_SIMD=AVX_256 -DGMX_GPU=ON -DCUDA_TOOLKIT_ROOT_DIR=/usr/lib/nvidia-cuda-toolkit/ -DCMAKE_INSTALL_PREFIX=/usr/local/ make -j2 sudo checkinstall
Если на каком-то шаге начинает ругаться об отсутствующих пакетах — ставим их. У меня ни на что не срыгнулась, хотя ставил на свежую ubuntu 14.04, так что все должно пройти «гладко».
Проверяем
Программа ругается на отсутствие входных файлов, что и ожидалось.
user@user-300E5C:~/source/gromacs-5.0.4/build$ grompp GROMACS: gmx grompp, VERSION 5.0.4 GROMACS is written by: ..... GROMACS: gmx grompp, VERSION 5.0.4 Executable: /usr/local/bin/gmx Library dir: /usr/local/share/gromacs/top Command line: grompp ------------------------------------------------------- Program grompp, VERSION 5.0.4 Source code file: /home/user/MODELING/source/gromacs-5.0.4/src/gromacs/fileio/futil.cpp, line: 545 File input/output error: grompp.mdp For more information and tips for troubleshooting, please check the GROMACS
Программа ругается на отсутствие входных файлов, что и ожидалось.
Подробнее о установке можно найти
Установка VMD
VMD это программа визуализации молекул, т.е. просмотра результатов моделирования. Итак регистрируемся на сайте и скачиваем нужную нам версию VMD, рекомендую с поддержкой GPU.
Распаковываем, меняем если нужно пути установки и ставим, не забывая поменять название пакета на vmd и указать версию, при использовании checkinstall
cd vmd 1.9.1 ./configure cd src sudo checkinstall
Будем считать что этого нам достаточно и приступим к моделированию.
Моделирование
Моделирование можно разбить на несколько этапов.
- Решить что будет моделироваться и с какой целью, отсюда будут отталкиваться параметры моделирования.
- Найти/сделать файл с координатами молекул/ы (gro pdb)
- Создание топологии молекулы — т.е. сделать описание связей итп. Фактически сказать пакету gromacs с чем он будет работать
- Запуск моделирования
- релаксация
- само моделирование
- Анализ
Точно по пунктам порбегаться в статье не буду, т.к допустим отличия релаксации и самого моделирования только в конфигурационном файле, а анализ дело специфическое и длинное…
Вода
Для наглядности попробуем смоделировать очень простую систему — воду. Координатный файл и файл топологии молекулы воды есть в силовых полях, которые вшиты в сам пакет. Т.к. моделирование чего-либо в воде производится достаточно часто, то существует несколько типов моделей воды и одна из лучших это tip4p-ew. Эту модель воды мы и будем использовать.
В папке громакса, уже есть отрелаксированная модель в 216 молекул воды, её и будем использовать.
:~/computate/$ mkdir water_sug water_sug/water water_sug/sug :~/computate/$ cd water_sug/water :~/computate/water_sug/water/$ cp /usr/local/share/gromacs/top/tip4p.gro h2o_216.gro :~/computate/water_sug/water/$ genconf -f h2o_216.gro -nbox 2 2 2 -o h2o_1728.gro
Была скопирована отрелаксированная вода, в 216 молекул, далее она была размножена в трех направлениях «на еще одну», получился итоговый файл в 1728 молекул воды. Метод genconf является методом громакса, его описание можно найти в документации.
Наглядно посмотрим за тем, что было сделано:
vmd h2o_216.gro
открывшаяся картинка, не очень не очень красива, сделаем по лучше, для этого откроем graphics->representation и выберем в «drawing method»: VDW

теперь посмотрим, на большую картину «большой» файл воды, отжав D в отображении прошлого и используя file->new molecule, и так же меняем тип отрисовки атомов:

Теперь её отрелаксируем(т.к. 8 блоков на стыках скорее всего не отрелаксированны): для этого потребуются файлы параметра моделирования и топология молекулы, т.е. файлы отражающие то, какие связи в молекуле существуют, какие силы и на что могут действовать итп. Использовать будем поле сил OPLS/AA.
Краткая теория
Для избегания возникновения граничных эффектов используется периодические боксы, т.е. на граниах система сталкивается сама с собой. Т.к. моделирование производится в какой-то среде, у которой есть температура, давление, то существуют методы привода к указанным параметрам. Это так называемые баростаты и термостаты, они бывают разных видов и используются на разных этапах моделирования.
Кулоновские сил распространяются на бесконечность, но при этом быстро спадают, потом используется так называемый радиус обрезания или верлет(подробнее в документации).
Для расчетов существует несколько разных алгоритмов, которые отличаются скоростью, и параметрами работы. Допустим некоторые алгоритмы не могут работать не сильно «дестабилизированных» системах, но при этом очень быстрые, когда система в некотором смысле стабильна. Другие же алгоритмы невероятно медлительны, но могут вывести систему из сильного отклонения в слабое итп.
Поля сил — это набор эмпирических параметров описывающих внутри и межмолекулярные взаимодействия, по сути это числа в лернард-джонсовское взимодействие(или морзе или...), а так же параметры связи в молекуле. Т.е. сила стягивания связи, её длина, сила удержания плоских и двугранных углов итп. Все эти вещи достаточно подробно описаны в документации, а так же легко найти их описания на русском.
Разработчики рекомендуют, если не известно что нужно — использовать поле opls/aa.
Кулоновские сил распространяются на бесконечность, но при этом быстро спадают, потом используется так называемый радиус обрезания или верлет(подробнее в документации).
Для расчетов существует несколько разных алгоритмов, которые отличаются скоростью, и параметрами работы. Допустим некоторые алгоритмы не могут работать не сильно «дестабилизированных» системах, но при этом очень быстрые, когда система в некотором смысле стабильна. Другие же алгоритмы невероятно медлительны, но могут вывести систему из сильного отклонения в слабое итп.
Поля сил — это набор эмпирических параметров описывающих внутри и межмолекулярные взаимодействия, по сути это числа в лернард-джонсовское взимодействие(или морзе или...), а так же параметры связи в молекуле. Т.е. сила стягивания связи, её длина, сила удержания плоских и двугранных углов итп. Все эти вещи достаточно подробно описаны в документации, а так же легко найти их описания на русском.
Разработчики рекомендуют, если не известно что нужно — использовать поле opls/aa.
Cоздаем файл описывающий топологию используемой системы:
topol.top
; Include forcefield parameters #include "oplsaa.ff/forcefield.itp" ;указываем какое поле использовать #include "oplsaa.ff/tip4p.itp" ; говорим где находится топология воды [ system ] water1728 ;название системы [ molecules ] SOL 1728 ;говорим сколько типов молекул в системе, которые указаны в топологии воды+название
Создаем папку для релаксации equi, в ней файл параметров моделирования следующего содержания:
grompp.mdp
Подробнее о параметрах моделирования можно почитать в документации. Количество используемых параметров может быть намного больше или меньше, зависит от требования условий. Используемые параметры, янечестно скопировал из примеров когда-то изучаемых мной курсов и для образовательных целей этого вполне хватит.
Документация к gromacs достаточно подробная и если кто-то решит этим плотно заниматься, документацию стоит досконально прочитать, чтобы понимать в каком случае какие методы использовать, допустим того же учета кулоновского взаимодействия.
; title = water ; a string ; Run parameters integrator = md ; leap-frog integrator nsteps = 5000 ; 10 ps dt = 0.002 ; 2 fs ; Output control nstxout = 0 ; save coordinates every 0 ps nstvout = 0 ; save velocities every 0 ps nstxtcout = 100 ; save coordinates in compressed format every 0.2 ps nstenergy = 100 ; save energies every 0.2 ps nstlog = 100 ; update log file every 0.2 ps ; Neighborsearching ns_type = grid ; search neighboring grid cels nstlist = 5 ; 10 fs rlist = 1.2 ; short-range neighborlist cutoff (in nm) rcoulomb = 1.2 ; short-range electrostatic cutoff (in nm) rvdw = 1.2 ; short-range van der Waals cutoff (in nm) DispCorr = EnerPres ; long range dispersion corrections for Energy and Pressure ; Electrostatics coulombtype = PME ; Particle Mesh Ewald for long-range electrostatics pme_order = 4 ; cubic interpolation fourierspacing = 0.12 ; grid spacing for FFT optimize_fft = yes ewald_rtol = 1.0e-5 ; Temperature coupling is on tcoupl = V-rescale ; modified Berendsen thermostat tc-grps = System ; two coupling groups - more accurate tau_t = 0.5 ; time constant, in ps ref_t = 300 ; reference temperature, one for each group, in K ; Pressure coupling is on pcoupl = Berendsen ; Pressure coupling on in NPT pcoupltype = isotropic ; uniform scaling of box vectors tau_p = 2.0 ; time constant, in ps ref_p = 1.0 ; reference pressure, in bar compressibility = 4.5e-5 ; isothermal compressibility of water, bar^-1 ; Periodic boundary conditions pbc = xyz ; 3-D PBC ; Velocity generation gen_vel = yes ; Velocity generation is off gen_temp = 300 ; initial temperature gen_seed = 2011 ; random seed
Подробнее о параметрах моделирования можно почитать в документации. Количество используемых параметров может быть намного больше или меньше, зависит от требования условий. Используемые параметры, я
Документация к gromacs достаточно подробная и если кто-то решит этим плотно заниматься, документацию стоит досконально прочитать, чтобы понимать в каком случае какие методы использовать, допустим того же учета кулоновского взаимодействия.
Запускаем релаксацию, из папки equi:
grompp -p ../topol.top -c ../h2o_1728.gro -f grompp.mdp mdrun -v
Чтобы просмотреть, что получилось необходимо произвести «перенос» атомов, которые ушли по граничным условиям в координатном файле моделирования, используя встроенную утилиту громакса trjconv. После чего открываем начальный файл координат vmd и подгружаем в него координаты:
trjconv -f traj_comp.xtc -o trajout.xtc -pbc mol #на вопрос отвечаем 0 vmd ../h2o_1728.gro
В vmd нажимаем правой кнопкой на h2o_1728.gro, выбираем load data into molecule и там уже выбираем файл trajout.xtc и жмем load. Если кому-то интересно что будет без trjconv, то может подгрузить исходный traj_comp.xtc.
Теперь используя прокрутку в vmd можно наблюдать за движением молекул в процессе моделирования.
По идеи для получения более-менее реальной модели необходимо произвести дальнейшее моделирование, поменяв термостат и баростат и увеличив время самого моделирования примерно в 5-10 раз. Но на воду саму по себе смотреть не очень интересно, куда интереснее будет поместит туда какое-то вещество, допустим сахар. Эта задача оказывается не только интереснее в плане наблюдения, но и в реализации, потому как топологию сахара придется создавать самим, а не брать готовую.
Сахар
Сахаров на самом деле всяких много, т.к. взять конкретный сахар цели нету, то берем первый понравившийся. Да и я особо уже не помню, чем они там по формулам различаются, фруктозы, сахарозы… Я нагуглил вот такой. Беру первый, как я понял это сахароза, посмотрим на него, уже стандартно в vmd, вот только отображение поставим CPK:

Теперь надо получить топологию данной молекулы, т.е. описать кто с кем связан и какие атомы соответствуют атомам в поле сил. Это можно сделать по-разному, регулярный способ мне не известен. Попробуем получить топологию из имеющихся уже данных — связи атомов в файле pdb и информации в поле силы для атомов.
В «хорошем» pdb файле уже описаны связи атомов, потому есть надежда что из него можно сконвертировать топлогию. И эта задача уже решена ранее, если порыться на сайте gromacs, то можно найти скрипты от участников, там и находится данная перловая утилита — topolgen-1.1.
используя её генерируем файл топологии:
perl topolgen_1.1.pl -f 1.pdb -o sugar.top
если посмотреть на файл топологии то можно увидеть, что на позиции type стоят просто числа, а должны быть названия типов атомов из того набора сил, который используется. Для того чтобы соотнести данную молекулу с полем сил, надо посмотреть какие типы атомов используются, сколько у них связей и что им соответствует в наборе OPLS/AA.
Для этого откроем в vmd снова молекулу, перейдем в representation, сверху нажмем «Create Rep» и сменим отображение на CPK, затем выберем вкладку selections. В Selected atoms удалим все имеющееся и введем serial 1, нажмем apply. Если посмотреть на отображение, то теперь один атом стал круглым, это и есть атом 1. Теперь мы можем соотнести атом с теми что имеются в наборе поля сил. Чтобы это сделать открываем файл поля сил atomtypes.atp, находящийся в /usr/local/share/gromacs/top/oplsaa.ff. И смотрим что подходит для атома углерода связанного с углеродом, кислородом и водородом, т.е. углерод в оксане. Такого там нету, потому будем просто искать подходящий по связям т.е. opls_193, далее меняем в vmd в selected atoms на 2 и ищем для второго атома, я выбрал opls_182 и так далее получаем в итоге файл топологии.
Так же включил поле сил, добавил название молекулы и сделал название основания.
приведена только измененная часть:
sugar.top
#include "oplsaa.ff/forcefield.itp"
[ moleculetype ]
; Name nrexcl
sugar 3
[ atoms ]
; nr type resnr residue atom cgnr charge mass typeB chargeB massB
1 opls_193 1 sugar C1 1 0.000
2 opls_182 1 sugar C2 1 0.000
3 opls_182 1 sugar C2 1 0.000
4 opls_182 1 sugar C2 0 0.000
5 opls_182 1 sugar C2 0 0.000
6 opls_182 1 sugar C3 0 0.000
7 opls_180 1 sugar O1 0 0.000
8 opls_078 1 sugar O2 0 0.000
9 opls_078 1 sugar O2 0 0.000
10 opls_078 1 sugar O2 0 0.000
11 opls_078 1 sugar O2 0 0.000
12 opls_180 1 sugar O1 0 0.000
13 opls_193 1 sugar C2 0 0.000
14 opls_182 1 sugar C2 0 0.000
15 opls_182 1 sugar C2 0 0.000
16 opls_182 1 sugar C1 0 0.000
17 opls_182 1 sugar C2 0 0.000
18 opls_182 1 sugar C2 0 0.000
19 opls_078 1 sugar O2 0 0.000
20 opls_180 1 sugar O1 0 0.000
21 opls_078 1 sugar O2 0 0.000
22 opls_078 1 sugar O2 0 0.000
23 opls_078 1 sugar O2 0 0.000
24 opls_185 1 sugar H1 0 0.000
25 opls_185 1 sugar H1 0 0.000
26 opls_185 1 sugar H1 0 0.000
27 opls_185 1 sugar H1 0 0.000
28 opls_185 1 sugar H1 0 0.000
29 opls_185 1 sugar H1 0 0.000
30 opls_185 1 sugar H1 0 0.000
31 opls_079 1 sugar H2 0 0.000
32 opls_079 1 sugar H2 0 0.000
33 opls_079 1 sugar H2 0 0.000
34 opls_079 1 sugar H2 0 0.000
35 opls_185 1 sugar H1 0 0.000
36 opls_185 1 sugar H1 0 0.000
37 opls_185 1 sugar H1 0 0.000
38 opls_185 1 sugar H1 0 0.000
39 opls_185 1 sugar H1 0 0.000
40 opls_185 1 sugar H1 0 0.000
41 opls_185 1 sugar H1 0 0.000
42 opls_079 1 sugar H2 0 0.000
43 opls_079 1 sugar H2 0 0.000
44 opls_079 1 sugar H2 0 0.000
45 opls_079 1 sugar H2 0 0.000
; в самом конце файла дописываем, количество используемых молекул:
[ molecules ]
; Compound #mols
sugar 1
Конвертируем координатный файл из pdb в gro, заодно помещая его в коробку 4 на 4 на 4:
editconf -f 1.pdb -box 4 4 4 -o sugar.gro
Приводим его в соответствие с топологией, подписывая атомы и ставя название основания:
sugar.gro
nat0011
45
1 C 1 2.056 1.948 1.876
1 C 2 2.210 1.948 1.876
1 C 3 2.263 2.092 1.876
1 C 4 2.198 2.168 1.759
1 C 5 2.045 2.158 1.775
1 C 6 1.972 2.236 1.663
1 O 7 2.007 2.022 1.767
1 O 8 1.831 2.222 1.680
1 O 9 2.259 1.880 1.992
1 O 10 2.405 2.093 1.861
1 O 11 2.241 2.305 1.763
1 O 12 2.007 2.003 1.996
1 C 13 1.964 1.922 2.103
1 C 14 1.997 1.993 2.236
1 C 15 1.941 1.908 2.352
1 C 16 1.791 1.883 2.329
1 C 17 1.811 1.900 2.096
1 C 18 1.772 1.828 1.966
1 O 19 1.632 1.798 1.969
1 O 20 1.770 1.820 2.205
1 O 21 1.741 1.799 2.434
1 O 22 1.963 1.974 2.477
1 O 23 2.138 2.007 2.251
1 H 24 2.022 1.845 1.866
1 H 25 2.246 1.897 1.786
1 H 26 2.236 2.141 1.970
1 H 27 2.228 2.122 1.665
1 H 28 2.015 2.199 1.871
1 H 29 2.002 2.196 1.566
1 H 30 1.998 2.341 1.669
1 H 31 1.806 2.129 1.674
1 H 32 2.232 1.787 1.990
1 H 33 2.446 2.046 1.934
1 H 34 2.224 2.348 1.679
1 H 35 2.015 1.826 2.100
1 H 36 1.950 2.091 2.237
1 H 37 1.993 1.812 2.353
1 H 38 1.737 1.979 2.332
1 H 39 1.760 1.996 2.100
1 H 40 1.828 1.734 1.958
1 H 41 1.794 1.891 1.881
1 H 42 1.607 1.753 1.889
1 H 43 1.752 1.842 2.519
1 H 44 1.917 2.059 2.477
1 H 45 2.174 2.061 2.180
4.00000 4.00000 4.00000
Теперь пробуем запустить моделирование, для упрощения и проверки возьмем тот же файл grompp.mdp, что и использовали для воды.
grompp -p sugar.top -c sugar.gro -f grompp.mdp
В ответ получаем сообщения об ошибках. Они нам говорят о том, что в поле сил нету внутримолекулярного взаимодействия для углов, при заданных типах атомов(файл ffbonded.itp). Это вполне разумно, потому как сбор молекулы производился вручную, из не соответствующих атомов(надо было оксан, а его нету), а какие больше подошли. Возможно некими хитрыми комбинациями и можно запустить моделирование, так чтобы атомы были «нужные», но это уже будет скорее всего неверно. Так же будет неточность в моделировании по причине отсутствия эффективного заряда на атомах, — это столбец charge в файле топологии. Естественный вопрос возникает, откуда брать все эти числа? Можно как-то из экспериментов, а можно и посчитать. Заряды бывает считают теоретически, на бумаге (малликен), но это не сильно точно, куда лучше методом квантовой химии, только считать одну молекулу а не группу. Квантовая химия позволяет найти и внутри молекулярное взаимодействие.
Квантомеханические расчеты производятся в различных программах
После не долгих поисков в репозитории ATB нашел сахарозу. Создадим папку ATB, куда и скачаем файлы топологии и расположения атомов(берем all-atoms). Получается ранее проделанные действия с сахаром были бесполезны, но целью их было показать построение топологии.
В итоге получили 2 файла sug.top и sug.pdb. Запустим моделирование на их основе и файла параметров моделирования, который использовали для воды – в файл топологии дописываем:
sug.top
добавлено в начало
И в конец:
#include "gromos54a7.ff/forcefield.itp" [ moleculetype ] ; Name nrexcl VH26 3
И в конец:
[ system ] ; Name Sugar [ molecules ] ; Compound #mols VH26 4
Поместим 4 молекулы в одну «коробку»:
echo -e "sugar \n0\n4 4 4">"sug_4.gro" gmx insert-molecules -f sug_4.gro -nmol 4 -ci sug.gro -o sug_4.gro
Возьмем файл параметров моделирования воды, увеличим время в 10 раз и запустим:
cp ~/computate/water_sug/water/equi/grompp.mdp . ; параметр nsteps делаем 50000 grompp -p sug.top -c sug_4.gro -f grompp.mdp mdrun -v trjconv -f traj_comp.xtc -o trajout.xtc -pbc mol ; уберем дефекты переноса на граничных условиях
Результаты можно посмотреть в vmd.
Вода + сахар
Вода для данного силового поля рекомендованная spc, она хуже чем tip4p, но не будем усложнять и так перегруженную статью, используем spc.
Совместим воду и сахар:
cd ../.. mkdir mix cd mix head -n5 /usr/local/share/gromacs/top/spc216.gro >spc.gro ; на 2й строке ставим количество атомов 3, и дописываем размер бокса 1 1 1. cp ../sug/ATB/sug.gro . cp ../sug/ATB/sug.top sug.itp ;убираем строку с #include, блоки [system] и [molecules] cp ~/computate/water_sug/water/equi/grompp.mdp . gmx insert-molecules -f sug_4.gro -nmol 512 -box 4 4 4 -ci spc.gro -o mix.gro
Создаем файл топологии для смешанной системы:
topol.top
; Include forcefield parameters #include "gromos54a7.ff/forcefield.itp" #include "gromos54a7.ff/spc.itp" #include "sug.itp" [ system ] Sug+SOL [ molecules ] VH26 4 sol 512
spc.gro
216H2O,WATJP01,SPC216,SPC-MODEL,300K,BOX(M)=1.86206NM,WFVG,MAR. 1984
3
1SOL OW 1 .230 .628 .113
1SOL HW1 2 .137 .626 .150
1SOL HW2 3 .231 .589 .021
1 1 1
Далее запускаем моделирование:
grompp -p topol.top -c mix.gro -f grompp.mdp mdrun -v
Скорее всего получим ошибку, вызванную тем, что система сильно далеко от равновесия. Для решения данной проблемы — уменьшим шаг моделирования в 2 раза, до 0,001ps (в файле grompp.mdp строчка dt = 0.002; 2 fs). Для наглядности, я промоделировал до времени в 300ps. Результат приведен ниже.
Для дальнейшего анализа можно использовать методы встроенные в gromacs такие как функции радиального распределения, энергии, температуры итп, подробнее можно найти в документации. На анализе я останавливаться не буду ибо статья и так уже по моим меркам большая.
Итог
В данной статье показаны установки gromacs и программы визуализации моделирования VMD, на примере нескольких систем показан запуск моделирования и проблема построения топологии… Прошу читателей простить мне не оптимальность некоторых вещей и возможно не большую ошибочность утверждений. В моем понимании для «старта» в области моделирования, показанного должно быть достаточно. Больше информации можно найти в документации и в рассылке пользователей gromacs, где крайне дружелюбно пользователи и авторы программы отвечают даже на самые странные вопросы. Спасибо прочитавшим, надеюсь кому-то это окажется полезным.

