Иногда перед некоторыми химиками может встать задача получить картинку с публикационным качеством, на которой будет молекула, и над каждой связью будет подписан её порядок. В этом посте, на примере кораннулена, мы познакомимся с простейшими (полуэмпирическими) квантово-химическими расчётами, визуализацией молекул, узнаем про порядки связей, и напишем питоновский скрипт, который будет генерировать из результатов наших расчётов картинку при помощи LaTeX-овского пакета TikZ картинку, которую уже почти-почти можно вставлять в статью. Всё это под катом :)
Глава 1. В которой у нас встают волосы дыбом
Вроде мы себе со школы представляем, что есть молекулы, в них могут быть одинарные, двойные, тройные связи, всё такое. Но на самом деле, все эти концепции верны только когда в образовании химической связи участвуют только два атома. В случае же более сложных химических связей (сопряжённые, трёхцентровые, и прочие извращённые), все эти понятия очень сильно размазываются.
Но всё равно, существуют способы каждую химическую связь охарактеризовать, приписав ей порядок связи. Грубо говоря, порядок связи между двумя атомами -- это число пар электронов, которые держат эту пару атомов вместе. Электроны описываются квантовой механикой, и в приниципе, решить задачу о квантовом состоянии электронов (в низшем по энергии состоянии) для любой сейчас не вызывает никаких сложностей. Зная же состояние всех электронов в молекуле (или электронную волновую функцию), мы можем применить её для расчёта порядка связи между каждой парой атомов (даже между несвязанными).
 и этин (ацетилен), как примеры молекул с одинарной, двойной и тройной связями.")
Если мы посчитаем все эти порядки связей, мы наверное захотим их визуализировать. И если с целыми порядками связей всё понятно (одинарная -- одна палочка между атомами, двойная -- две, тройная -- три, см. рисунок выше), то что делать в случае, если порядок связи равен 1.3, уже не так очевидно. Поэтому самым простым решением было бы изобразить все связи в молекулах черточками, а порядок связи подписать сверху.
Но каким бы очевидным это всё ни было для нас, мало какой визуализатор молекул с такой задачей справится. Из тех, что я знаю, это только GaussView, который, за несколько тысяч у.е. своей стоимости, сможет построить абсолютно некрасивую и непубликабельную картинку, так что этот вариант мы отклоняем. Вместо этого, мы попробуем сами сгенерировать картинку в читаемом варианте при помощи LaTeX-овского безумно мощного пакета TikZ.
Глава 2. В которой мы знакомимся с визуализацией молекул
Чтобы что-то построить, нам надо выбрать молекулу, и что-то с ней сделать. Более интересными вариантами должны быть достаточно большие молекулы (чтобы имело смысл вообще заморачиваться со скриптами) и с достаточно извратными связями, например сопряжённые системы.

От этого всего, у меня в голове от этого всплыл класс молекул, называемых полициклические ароматические углеводороды (ПАУ). Что же означает это название?
"Полициклические" -- значит они содержат в своей структуре несколько циклов, например, циклогексана, циклогексена, или бензола.
"Ароматические" означает, что хотя бы один из циклов должен быть сопряжённым так успешно, что образовалась бы ароматическая система, такая как в бензоле. Что такое ароматичность объяснить в двух словах очень сложно (несмотря на попытки школьного курса это сделать), и откровенно говоря, это определение ароматичности -- это один из священных граалей современной химии. Почитать об этом поподробнее можно, например, в этой статье.
"Углеводороды" же -- это самое простое. Это значит, что молекулы этого класса состоят из двух типов атомов: углерод (C) и водород (H).
Этот класс соединений очень важен как на Земле (ибо является одним из наиболее стрёмных загрязнителей воды и воздуха), так и под землёй (как один из заметных компонентов нефти), и даже давным-давно в одной далёкой-предалёкой галактике в космическом пространстве, где молекулы этого класса, по последним оценкам, ответственны за 10% всего запаса галактического углерода.

В качестве нашей пробной молекулы, выберем кораннулен (C20H10). Это очень красивая молекула, которая представляет собой пять бензольных колец, присоединённых, как лепестки цветка, к пятичленному циклу. Из-за стерического напряжения, эта молекула плоская, и напоминает чашечку. И в эту чашечку даже наливали воду (в количестве одной молекулы, см. картинку выше, выкусите, гомеопаты!).
Чтобы нам визуализировать молекулу, а после чего посчитать её электронную структуру, нам нужны координаты каждого атома. Для этого мы можем воспользоваться или программами-построителями молекулярных геометрий (например, Кемкрафтом, или Jmol-ом), или просто найти эти координаты в интернете. Неплохо можно разжиться на сайте NIST Chemistry Webbook. Почти для каждой молекулы в базе данных, там есть ссылка на скачивание "computed 3d SD file". Тыкая туда, мы скачаем файл в формате SDF. Файл по поиску "corannulene" дан тут:
файл 5821-51-2-3d.sdf
его скачали с NIST Chemistry Webbook
NIST 08021410203D 1 1.00000 -768.14928 Copyright by the U.S. Sec. Commerce on behalf of U.S.A. All rights reserved. 30 35 0 0 0 0 0 0 0 0999 V2000 2.3479 2.7598 -0.4735 C 0 0 0 0 0 0 0 0 0 0 0 0 2.3108 4.1732 -0.5616 C 0 0 0 0 0 0 0 0 0 0 0 0 3.6394 4.6362 -0.7268 C 0 0 0 0 0 0 0 0 0 0 0 0 4.4974 3.5091 -0.7410 C 0 0 0 0 0 0 0 0 0 0 0 0 3.6992 2.3494 -0.5843 C 0 0 0 0 0 0 0 0 0 0 0 0 1.3863 2.0195 0.1935 C 0 0 0 0 0 0 0 0 0 0 0 0 1.3101 4.9394 0.0122 C 0 0 0 0 0 0 0 0 0 0 0 0 4.1781 1.1717 -0.0353 C 0 0 0 0 0 0 0 0 0 0 0 0 5.8271 3.5678 -0.3589 C 0 0 0 0 0 0 0 0 0 0 0 0 4.0547 5.8962 -0.3297 C 0 0 0 0 0 0 0 0 0 0 0 0 0.2292 2.7852 0.6078 C 0 0 0 0 0 0 0 0 0 0 0 0 0.1931 4.1718 0.5217 C 0 0 0 0 0 0 0 0 0 0 0 0 1.6744 6.3236 0.2317 C 0 0 0 0 0 0 0 0 0 0 0 0 2.9776 6.7779 0.0694 C 0 0 0 0 0 0 0 0 0 0 0 0 5.4848 6.0167 -0.1373 C 0 0 0 0 0 0 0 0 0 0 0 0 6.3264 4.9110 -0.1513 C 0 0 0 0 0 0 0 0 0 0 0 0 1.8210 0.6936 0.5807 C 0 0 0 0 0 0 0 0 0 0 0 0 3.1467 0.2910 0.4720 C 0 0 0 0 0 0 0 0 0 0 0 0 5.6119 1.1509 0.1665 C 0 0 0 0 0 0 0 0 0 0 0 0 6.3949 2.2886 0.0128 C 0 0 0 0 0 0 0 0 0 0 0 0 -0.6061 2.2758 1.0834 H 0 0 0 0 0 0 0 0 0 0 0 0 -0.6692 4.6919 0.9333 H 0 0 0 0 0 0 0 0 0 0 0 0 0.9312 7.0163 0.6204 H 0 0 0 0 0 0 0 0 0 0 0 0 3.2019 7.8080 0.3376 H 0 0 0 0 0 0 0 0 0 0 0 0 5.9103 6.9859 0.1137 H 0 0 0 0 0 0 0 0 0 0 0 0 7.3769 5.0594 0.0892 H 0 0 0 0 0 0 0 0 0 0 0 0 1.1133 0.0161 1.0532 H 0 0 0 0 0 0 0 0 0 0 0 0 3.4230 -0.6854 0.8639 H 0 0 0 0 0 0 0 0 0 0 0 0 6.0864 0.2438 0.5345 H 0 0 0 0 0 0 0 0 0 0 0 0 7.4508 2.2264 0.2670 H 0 0 0 0 0 0 0 0 0 0 0 0 2 1 1 0 1 5 1 0 5 4 1 0 4 3 1 0 2 3 1 0 2 7 2 0 7 12 1 0 12 11 2 0 11 6 1 0 1 6 2 0 4 9 2 0 9 16 1 0 16 15 2 0 15 10 1 0 3 10 2 0 5 8 2 0 8 19 1 0 19 20 2 0 9 20 1 0 6 17 1 0 17 18 2 0 8 18 1 0 10 14 1 0 14 13 2 0 7 13 1 0 11 21 1 0 12 22 1 0 13 23 1 0 14 24 1 0 15 25 1 0 16 26 1 0 17 27 1 0 18 28 1 0 19 29 1 0 20 30 1 0 M END > <COPYRIGHT> Collection (C) 2016 copyright by the U.S. Secretary of Commerce on behalf of the United States of America. All rights reserved. > <DATE> 2014-08-02 > <CAS.NUMBER> 5821-51-2 > <METHOD> B3LYP/6-31G* > <DIPOLE.MOMENT> 1.7256 debye > <ELECTRONIC.ENERGY> -768.149281981 hartree > <IR.FREQUENCIES> "Frequency (cm-1)" "Intensity (km/mol)" 140.9463 4.3688 142.1225 0.0234 142.4048 0.0012 282.0224 0.0000 282.2456 0.0000 311.8107 0.2249 312.3830 0.2255 409.5691 2.3881 409.7950 2.3915 438.7769 0.0000 438.8373 0.0000 452.9639 3.4550 453.1293 3.4725 542.0487 0.0001 547.5829 0.0002 547.6385 0.0000 559.9977 13.1405 605.0577 0.8047 612.4161 0.0001 612.5322 0.0000 648.1763 0.0001 648.4241 0.0001 649.6040 0.0000 673.0974 16.0743 673.1653 16.0396 759.3405 3.7537 759.4716 3.7365 772.4206 0.0000 772.7373 0.0000 812.4235 0.0001 812.6339 0.0000 833.7793 3.4835 833.8699 3.4724 856.2070 111.8882 869.7445 0.0162 869.8674 0.0150 937.3935 0.0000 953.5278 0.0002 960.1158 0.4483 960.1906 0.4495 968.6705 0.0001 968.8939 0.0004 1051.9088 1.2196 1091.4120 0.0000 1091.5541 0.0000 1171.6836 2.1672 1171.8624 3.5197 1171.9526 1.3649 1172.0671 1.7608 1197.1583 0.0000 1197.5385 0.0000 1224.1857 0.0001 1224.4120 0.0001 1244.7627 0.0000 1269.6389 1.0906 1342.9936 9.2123 1343.2742 9.1932 1391.6399 0.0001 1391.8987 0.0000 1440.8503 0.0001 1440.8972 0.0000 1458.5640 1.8664 1458.7683 1.8785 1480.2637 1.8568 1486.1287 2.7000 1486.1836 2.7010 1497.5004 0.0001 1497.7884 0.0001 1527.1617 0.0000 1668.3089 0.0041 1668.4748 0.0110 1669.7603 1.0670 1669.9892 1.0731 1671.5673 0.0030 3174.2046 0.3130 3174.9229 5.3562 3175.0279 5.6321 3175.6846 0.3267 3175.8603 0.0159 3191.4230 1.7604 3191.6544 0.1704 3192.6984 100.6297 3192.7960 102.6391 3194.2245 6.6723 > <ROTATIONAL.CONSTANTS> 0.50785 GHz 0.50779 GHz 0.26288 GHz > <SOFTWARE> Gaussian 09, Revision D.01 > <CONTRIBUTOR> Ethan Ho $$$$
Там находятся координаты всех атомов молекулы, данные в ангстремах (1 Å = 10-10 м). Этот файл мы можем легко визуализировать любым молекулярным вьюером (хороший список есть в Википедии). Я это люблю делать в Jmol, упомянутом выше. Команда jmol 5821-51-2-3d.sdf & позволяет мне узреть сию красоту:

Всю эту молекулку можно теперь сохранять, покрутить и т.д. и т.п., сохранив в нужной конфигурации, используя кнопочкуmodel kit. Заметим, что тут у нас изображены порядки связей (одинарные и двойные черточки), что является результатом того, что они были в нашем изначальном SDF-файле.
После визуализации, выберем удобную для нас ориентацию, т.к. мы будем проецировать молекулу на плоскость xy (что мы и видим в Jmol). После чего вытащим руками геометрию молекулы в XYZ формате, и сохраним её в файл ini.xyz:
файл ini.xyz со структурой молекулы
30 C 1.69979 2.78101 -1.65228 C 1.35394 3.94963 -2.37448 C 2.55242 4.62236 -2.71788 C 3.63875 3.86951 -2.20816 C 3.11178 2.73154 -1.54944 C 0.85441 2.18448 -0.73191 C 0.14026 4.59911 -2.22325 C 3.77155 2.08239 -0.51937 C 4.86014 4.43362 -1.88024 C 2.61606 5.98885 -2.93335 C -0.48972 2.72307 -0.72415 C -0.82877 3.86975 -1.43236 C 0.14917 5.98162 -2.65393 C 1.32473 6.64151 -2.99106 C 3.94461 6.54944 -2.80136 C 5.01022 5.81082 -2.30138 C 1.52592 1.32553 0.22109 C 2.91115 1.27701 0.32197 C 5.13459 2.52641 -0.31466 C 5.65151 3.64279 -0.96086 H -1.24117 2.27461 -0.07791 H -1.83214 4.27256 -1.31201 H -0.77553 6.55407 -2.63124 H 1.27275 7.70397 -3.21872 H 4.10291 7.60441 -3.01443 H 5.95982 6.31750 -2.14339 H 0.93627 0.76426 0.94247 H 3.34968 0.67971 1.11834 H 5.75177 2.02999 0.43104 H 6.65244 3.97556 -0.69475
Этот файл мы всё также можем открывать Jmol-ом, или другим просмотрщиком молекул, но он нам понадобится для квантово-химических расчётов, т.е. для нахождения наиболее выгодной конфигурации электронов и их энергии.
Глава 3. В которой мы проводим квантово-химический расчёт
Итак, мы создали XYZ-файл ini.xyz, содержащий изначальные координаты молекулы. Теперь нам бы эту молекулу оптимизировать (т.е. найти оптимальное расположение всех атомов относительно друг друга, которое будет минимизировать энергию электронов, т.е. всех химических связей). Для этого мы можем воспользоваться программой XTB. Это не полновесная квантовая химия, а т.н. полуэмпирика, т.е. квантово-механический расчёт там происходит, но некоторые вещи хитренько запараметризованы. Подобные методы нужны в первую очередь для ускорения вычислений, что позволяет вычислять параметры гигантских систем в тысячи атомов.
После установки (по-сути прописывания пути к бинарнику) провести расчёт в XTB проще пареной репы:
Запускаем команду
xtb ini.xyz --ohess > output.logЖдём
Получаем результат
Заклинание "ohess" означет "optimization + hessian". Первое -- это реально оптимизация энергии системы как функции координат атомов, а второе -- это расчёт колебательных частот молекулы. Да, молекула колеблется, и чтобы узнать как это она делает, мы можем посчитать вторые производные (Гессиан) её энергии в точке минимума и в таком эффективном потенциале найти формы и частоты колебаний атомов. Полезная штука, которой мы не воспользуемся (но желающие могут их посмотреть в том же Jmol-е, открыв полученный файл g98.out).
Из кучи всяких файлов нас интересуют несколько. xtbopt.xyz -- это там, где содержится оптимизированная геометрия молекулы, минимум на электронной поверхности потенциальной энергии. Этот файл мы переименуем в mol.xyz (вот он под спойлером).
файл mol.xyz
30 energy: -47.909984727343 gnorm: 0.000561323830 xtb: 6.4.1 (afa7bdf) C 1.70342497008918 2.77120382029981 -1.67459989574751 C 1.35888198845737 3.93605651904526 -2.39411238929612 C 2.55320713127695 4.60662002508876 -2.73648638740711 C 3.63588083564752 3.85619478604766 -2.22858426799746 C 3.11068612200065 2.72184425174820 -1.57229992069963 C 0.86531732896573 2.19467015713543 -0.73988026274033 C 0.15592822786286 4.59298913710542 -2.22131034952792 C 3.76274802514278 2.09303373963485 -0.52926100615237 C 4.84408005491831 4.42856531897689 -1.88049627334238 C 2.61494436161795 5.97362661868611 -2.92622049178063 C -0.46023666409747 2.73788915472265 -0.72902661742635 C -0.79568990271412 3.87198196823774 -1.42956257287266 C 0.17248681723255 5.96311018441904 -2.63939045084267 C 1.33528769236052 6.61597608493287 -2.97271643766150 C 3.92951616360376 6.52499048543271 -2.78412401179422 C 4.98361507251879 5.79437063530864 -2.28963603342726 C 1.53562915869062 1.35451068920168 0.20727845907322 C 2.90574505520454 1.30644138413572 0.30686703629669 C 5.10743028420101 2.54260766563073 -0.32428692794655 C 5.61876291181198 3.64701225551270 -0.96325052396703 H -1.19874170458545 2.29409153379029 -0.07494466776196 H -1.78764656957806 4.28504174115813 -1.30476343298049 H -0.74868879805596 6.52904857077271 -2.60304052043814 H 1.29265341328483 7.67518070709473 -3.18821270199824 H 4.07652658282649 7.57755348473410 -2.98578625864465 H 5.92703793057654 6.29492483888218 -2.11767956737328 H 0.94303089560637 0.80961584278484 0.92983205611095 H 3.34832686183346 0.72523869909287 1.10467238401189 H 5.71098816523577 2.05170297389314 0.42731147860243 H 6.60865448964361 3.99053882857075 -0.69441367658131
То, куда мы редиректнули всю выдачу (output.log) -- это собственно лог программы, там много всякой полезной и бесполезной (нам) инфы. Например, там есть энергии орбиталей, частоты колебаний, термодинамические параметры молекулы и т.д. Но что мы хотим -- это порядки связей, вытащенные из электронной плотности. XTB вычисляет т.н. порядки связей Уибера (Wiber bond orders, WBO), которые были введены в работе 1966-го года. Ищем в логе последний кусок такого вида:
Wiberg/Mayer (AO) data. largest (>0.10) Wiberg bond orders for each atom --------------------------------------------------------------------------- # Z sym total # sym WBO # sym WBO # sym WBO --------------------------------------------------------------------------- 1 6 C 3.984 -- 6 C 1.345 2 C 1.192 5 C 1.192 2 6 C 3.984 -- 7 C 1.345 3 C 1.192 1 C 1.192 3 6 C 3.984 -- 10 C 1.345 2 C 1.192 4 C 1.192 4 6 C 3.984 -- 9 C 1.345 5 C 1.192 3 C 1.192 5 6 C 3.984 -- 8 C 1.345 4 C 1.192 1 C 1.192 6 6 C 3.986 -- 1 C 1.345 17 C 1.220 11 C 1.220 7 6 C 3.986 -- 2 C 1.345 13 C 1.220 12 C 1.220 8 6 C 3.986 -- 5 C 1.345 19 C 1.220 18 C 1.220 9 6 C 3.986 -- 4 C 1.345 20 C 1.220 16 C 1.220 10 6 C 3.986 -- 3 C 1.345 14 C 1.220 15 C 1.220 11 6 C 3.980 -- 12 C 1.624 6 C 1.220 21 H 0.969 12 6 C 3.980 -- 11 C 1.624 7 C 1.220 22 H 0.969 13 6 C 3.980 -- 14 C 1.623 7 C 1.220 23 H 0.969 14 6 C 3.980 -- 13 C 1.623 10 C 1.220 24 H 0.969 15 6 C 3.980 -- 16 C 1.624 10 C 1.220 25 H 0.969 16 6 C 3.980 -- 15 C 1.624 9 C 1.220 26 H 0.969 17 6 C 3.980 -- 18 C 1.624 6 C 1.220 27 H 0.969 18 6 C 3.980 -- 17 C 1.624 8 C 1.220 28 H 0.969 19 6 C 3.980 -- 20 C 1.623 8 C 1.220 29 H 0.969 20 6 C 3.980 -- 19 C 1.623 9 C 1.220 30 H 0.969 21 1 H 0.998 -- 11 C 0.969 22 1 H 0.998 -- 12 C 0.969 23 1 H 0.998 -- 13 C 0.969 24 1 H 0.998 -- 14 C 0.969 25 1 H 0.998 -- 15 C 0.969 26 1 H 0.998 -- 16 C 0.969 27 1 H 0.998 -- 17 C 0.969 28 1 H 0.998 -- 18 C 0.969 29 1 H 0.998 -- 19 C 0.969 30 1 H 0.998 -- 20 C 0.969 ---------------------------------------------------------------------------
Собственно, столбцы WBO нам и нужны! Вырезаем их вместе с парами атомов (столбцы #) в отдельный файл "bonds.dat" в формате "<номер первого атома> <номер второго атома> <WBO этой пары атомов>":
файл bonds.dat
1 6 1.345 2 7 1.345 3 10 1.345 4 9 1.345 5 8 1.345 6 1 1.345 7 2 1.345 8 5 1.345 9 4 1.345 10 3 1.345 11 12 1.624 12 11 1.624 13 14 1.624 14 13 1.624 15 16 1.624 16 15 1.624 17 18 1.624 18 17 1.624 19 20 1.624 20 19 1.624 21 11 0.969 22 12 0.969 23 13 0.969 24 14 0.969 25 15 0.969 26 16 0.969 27 17 0.969 28 18 0.969 29 19 0.969 30 20 0.969 1 5 1.192 2 3 1.192 3 4 1.192 4 3 1.192 5 1 1.192 6 17 1.220 7 12 1.220 8 18 1.220 9 16 1.220 10 15 1.220 11 6 1.220 12 7 1.220 13 7 1.220 14 10 1.220 15 10 1.220 16 9 1.220 17 6 1.220 18 8 1.220 19 8 1.220 20 9 1.220 1 2 1.192 2 1 1.192 3 2 1.192 4 5 1.192 5 4 1.192 6 11 1.220 7 13 1.220 8 19 1.220 9 20 1.220 10 14 1.220 11 21 0.969 12 22 0.969 13 23 0.969 14 24 0.969 15 25 0.969 16 26 0.969 17 27 0.969 18 28 0.969 19 29 0.969 20 30 0.969
И после всей этой подготовке можно приступать к построению картинки.
Глава 4. В которой мы генерируем TikZ-картинку
Собственно, имея файлы "mol.xyz" и "bonds.dat", мы запускаем скрипт make_tikz_picture_bond_orders.py из-под спойлера, и получаем наш код.
скрипт make_tikz_picture_bond_orders.py
#! /usr/bin/python import numpy as np # сначала прочитаем файл с молекулярной геометрией inpf = open("mol.xyz", "r") # открываем файл 'mol.xyz' в "read" режиме count=0 # это просто счётчик строк xyz = [] # здесь мы будем хранить x,y,z, координаты атомов atn = [] # а здесь тип атома for line in inpf: # идём по каждой строке файла count+=1 # не забывая повышать счётчик # первые 2 строки xyz-файла -- это число атомов и строка с описанием, # их мы пропускам, т.к. координаты начинаются с 3й строки if count>2: words = line.split() # делим строку на слова if len(words)>=4: # не обязательно, но вдруг что-то не так с файлом # первое слово в строке -- тип атома, # переводим его заодно в строчечные буквы, чтобы # избежать разночтений atn.append(words[0].lower()) # ну а потом остальные слова (координаты в ангстремах) # сохраняем в лист, переводя их floats xyz.append([float(word) for word in words[1:4]]) inpf.close() # ну и закрываем файл # теперь читаем файл со связями и их порядками inpf = open("bonds.dat", "r") # открываем файл 'bonds.dat' в "read" режиме BondInd = [] # здесь мы будем хранить индексы атомов, между которыми есть связь BondOrd = [] # а здесь численное значение для связи (предположительно, порядок связи) for line in inpf: # идём по каждой строке файла words = line.split() # делим строку на слова if len(words)>=3: # не обязательно, но вдруг что-то не так с файлом # индексы переводим в numpy.array, конвертируя str в int # и убирая лишнюю единицу, т.к. в программах файлы нумеруют с 1, # а у нас здесь индексы идут с 0 BondInd.append(np.array(words[:2], dtype=int)-1) # а численное значение сохраняем отдельно, конвертируя str во float BondOrd.append(float(words[2])) inpf.close() # ну и закрываем файл # открываем файл, куда будем всё писать, назовём его 'fig.tex' outf = open("fig.tex", "w") # это функция для строки, которая рисует связь в TikZ # берёт два индекса, номера атомов из BondInd[n], i и j, # а также значение порядка связи (bond order = bo). def DrawBond(i,j,bo): res = '\draw [ultra thick] ' res += ' (%7.4f,%7.4f) -- ' % (xyz[i][0],xyz[i][1]) # начало вектора res += ' (%7.4f,%7.4f) ' % (xyz[j][0],xyz[j][1]) # конец вектора res += ' node [pos=0.5,below] {%7.1f};' % (bo) # и подпись return res # Эти штуки мы считаем, чтобы получить перспективу # берём среднее значение координаты z, чтобы # получить референсное значение для перспективы AvZ = np.mean([r[2] for r in xyz]) # и считаем амплитуду для координаты Z ZAmp = np.abs(max([r[2] for r in xyz]) - min([r[2] for r in xyz])) # молекула может быть плоской и вся лежать в плоскости XY, # поэтому если изменение координаты Z слишком мало, то делить на околонулевое # значение, наверное, не стоит. Поэтому ставим такой нижний порог на амплитуду, # в 0.5 ангстрем ZAmp = max(0.5,ZAmp) # а это функция для рисования атома в виде шарика, # нам нужен только индекс атома, i def DrawAtom(i): res = '\draw' if atn[i] == 'c': # это мы рисуем углерод Rad = 0.7 # ковалентный радиус углерода, в ангстремах res += '[atomC] ' elif atn[i] == 'h': # это мы рисуем углерод Rad = 0.25 # ковалентный радиус водорода, в ангстремах res += '[atomH] ' else: # а это для не заданных атомов, какие-то рандомные настройки Rad = 0.5 res += '[atomX3] ' # позиция атома -- это проекция на плоскось xy x = xyz[i][0] # x-координата y = xyz[i][1] # y-координата # а это радиус атома на картинке, который зависит от перспективы r = Rad * 0.3 * (1.0 + 0.9 * (xyz[i][2] - AvZ)/ZAmp) res += '(%7.4f,%7.4f) circle (%.3f);' % (x,y, r) return res # собственно, это 'шапка' LaTeX-овского документа # с настройками TikZ # здесь мы задаём параметры для атомов C и H, но их можно и # надобавлять других HeadFile=r"""\documentclass{article} \usepackage[utf8]{inputenc} \usepackage{tikz} \usepackage{xcolor} \usepackage{color} \begin{document} \begin{center} \begin{tikzpicture}[ >=stealth, atomC/.style={shade, ball color=gray!50!black}, atomH/.style={shade, ball color=white!50!gray}, atomX3/.style={shade, ball color=orange!50!white}, scale=1. ] """ # а это низ LaTeX-овского документа TailFile=''' \end{tikzpicture} \end{center} \end{document} ''' # теперь начинаем запись LaTeX-овского документа outf.write(HeadFile) # начинаем с шапки # потом идём по всем связям for n,bo in enumerate(BondOrd): # при помощи функции DrawBond получаем # TikZ команду для рисования связи line = DrawBond(BondInd[n][0],BondInd[n][1],bo) outf.write(line+"\n") # а теперь рисуем поверх шарики атомов for n in range(0,len(atn)): line = DrawAtom(n) outf.write(line+"\n") # не забываем низ файла. outf.write(TailFile) outf.close() # и закрыть файл
Этот монстр создаст файл fig.tex из файлов mol.xyz и bonds.dat.
Содержимое этого кода тупо, банально и плохо написано, но всё же прокомментируем что он делает.
Сначала он читает геометрию молекулы и связи в этой молекулы, заданные в наших файлах
mol.xyzиbonds.dat. Складируем это всё в несколько списков.Выкатывем шапку LaTeX-овского документа в файл
fig.tex, которые и является нашим искомым результатом.Начинаем генерировать команды построения связей, каркас молекулы. Для этого у нас есть команды TikZ вида
\draw [ultra thick] (x1,y1) -- (x2,y2) node [pos=0.5,below] {WBO};Эта команда рисует линию от точки(x1,y1)к точке(x2,y2), а ещё посередине линии (pos=0.5-- это позиция на половине, снизу) выдаст надписьWBO. В принципе, этого достаточно уже для наших целей, но в целях дополнительного украшательства есть следующий шаг.Сверху позиций атомов нарисуем шарики, да не просто так, а с перспективой. Для этого нам нужна команда вида
\draw[atomType] (x,y) circle (r);atomType-- это стиль шарика, который мы пре-задали в шапке LaTeX-овского документа, поскольку у нас только углерод и водород, их только мы и определили.(x,y)-- это позиция центра шара (проекция молекулы на плоскость xy). Ну аr-- это радиус шара, с помощью которого мы создаём перспективу, отображая осьzна рисунке. То, как мы вычисляем этот радиус поговорим ниже.Ну и в конце добавляем "хвост" LaTeX-овского документа, чтобы он у нас вообще скомпилировался.
В результате мы получаем LaTeX-овский код в файле fig.tex с картинкой, генерируемой пакетом TikZ.
полученный LaTeX-овский код
\documentclass{article} \usepackage[utf8]{inputenc} \usepackage{tikz} \usepackage{xcolor} \usepackage{color} \begin{document} \begin{center} \begin{tikzpicture}[ >=stealth, atomC/.style={shade, ball color=gray!50!black}, atomH/.style={shade, ball color=white!50!gray}, atomX3/.style={shade, ball color=orange!50!white}, scale=1. ] \draw [ultra thick] ( 1.7034, 2.7712) -- ( 0.8653, 2.1947) node [pos=0.5,below] { 1.3}; \draw [ultra thick] ( 1.3589, 3.9361) -- ( 0.1559, 4.5930) node [pos=0.5,below] { 1.3}; \draw [ultra thick] ( 2.5532, 4.6066) -- ( 2.6149, 5.9736) node [pos=0.5,below] { 1.3}; \draw [ultra thick] ( 3.6359, 3.8562) -- ( 4.8441, 4.4286) node [pos=0.5,below] { 1.3}; \draw [ultra thick] ( 3.1107, 2.7218) -- ( 3.7627, 2.0930) node [pos=0.5,below] { 1.3}; \draw [ultra thick] ( 0.8653, 2.1947) -- ( 1.7034, 2.7712) node [pos=0.5,below] { 1.3}; \draw [ultra thick] ( 0.1559, 4.5930) -- ( 1.3589, 3.9361) node [pos=0.5,below] { 1.3}; \draw [ultra thick] ( 3.7627, 2.0930) -- ( 3.1107, 2.7218) node [pos=0.5,below] { 1.3}; \draw [ultra thick] ( 4.8441, 4.4286) -- ( 3.6359, 3.8562) node [pos=0.5,below] { 1.3}; \draw [ultra thick] ( 2.6149, 5.9736) -- ( 2.5532, 4.6066) node [pos=0.5,below] { 1.3}; \draw [ultra thick] (-0.4602, 2.7379) -- (-0.7957, 3.8720) node [pos=0.5,below] { 1.6}; \draw [ultra thick] (-0.7957, 3.8720) -- (-0.4602, 2.7379) node [pos=0.5,below] { 1.6}; \draw [ultra thick] ( 0.1725, 5.9631) -- ( 1.3353, 6.6160) node [pos=0.5,below] { 1.6}; \draw [ultra thick] ( 1.3353, 6.6160) -- ( 0.1725, 5.9631) node [pos=0.5,below] { 1.6}; \draw [ultra thick] ( 3.9295, 6.5250) -- ( 4.9836, 5.7944) node [pos=0.5,below] { 1.6}; \draw [ultra thick] ( 4.9836, 5.7944) -- ( 3.9295, 6.5250) node [pos=0.5,below] { 1.6}; \draw [ultra thick] ( 1.5356, 1.3545) -- ( 2.9057, 1.3064) node [pos=0.5,below] { 1.6}; \draw [ultra thick] ( 2.9057, 1.3064) -- ( 1.5356, 1.3545) node [pos=0.5,below] { 1.6}; \draw [ultra thick] ( 5.1074, 2.5426) -- ( 5.6188, 3.6470) node [pos=0.5,below] { 1.6}; \draw [ultra thick] ( 5.6188, 3.6470) -- ( 5.1074, 2.5426) node [pos=0.5,below] { 1.6}; \draw [ultra thick] (-1.1987, 2.2941) -- (-0.4602, 2.7379) node [pos=0.5,below] { 1.0}; \draw [ultra thick] (-1.7876, 4.2850) -- (-0.7957, 3.8720) node [pos=0.5,below] { 1.0}; \draw [ultra thick] (-0.7487, 6.5290) -- ( 0.1725, 5.9631) node [pos=0.5,below] { 1.0}; \draw [ultra thick] ( 1.2927, 7.6752) -- ( 1.3353, 6.6160) node [pos=0.5,below] { 1.0}; \draw [ultra thick] ( 4.0765, 7.5776) -- ( 3.9295, 6.5250) node [pos=0.5,below] { 1.0}; \draw [ultra thick] ( 5.9270, 6.2949) -- ( 4.9836, 5.7944) node [pos=0.5,below] { 1.0}; \draw [ultra thick] ( 0.9430, 0.8096) -- ( 1.5356, 1.3545) node [pos=0.5,below] { 1.0}; \draw [ultra thick] ( 3.3483, 0.7252) -- ( 2.9057, 1.3064) node [pos=0.5,below] { 1.0}; \draw [ultra thick] ( 5.7110, 2.0517) -- ( 5.1074, 2.5426) node [pos=0.5,below] { 1.0}; \draw [ultra thick] ( 6.6087, 3.9905) -- ( 5.6188, 3.6470) node [pos=0.5,below] { 1.0}; \draw [ultra thick] ( 1.7034, 2.7712) -- ( 3.1107, 2.7218) node [pos=0.5,below] { 1.2}; \draw [ultra thick] ( 1.3589, 3.9361) -- ( 2.5532, 4.6066) node [pos=0.5,below] { 1.2}; \draw [ultra thick] ( 2.5532, 4.6066) -- ( 3.6359, 3.8562) node [pos=0.5,below] { 1.2}; \draw [ultra thick] ( 3.6359, 3.8562) -- ( 2.5532, 4.6066) node [pos=0.5,below] { 1.2}; \draw [ultra thick] ( 3.1107, 2.7218) -- ( 1.7034, 2.7712) node [pos=0.5,below] { 1.2}; \draw [ultra thick] ( 0.8653, 2.1947) -- ( 1.5356, 1.3545) node [pos=0.5,below] { 1.2}; \draw [ultra thick] ( 0.1559, 4.5930) -- (-0.7957, 3.8720) node [pos=0.5,below] { 1.2}; \draw [ultra thick] ( 3.7627, 2.0930) -- ( 2.9057, 1.3064) node [pos=0.5,below] { 1.2}; \draw [ultra thick] ( 4.8441, 4.4286) -- ( 4.9836, 5.7944) node [pos=0.5,below] { 1.2}; \draw [ultra thick] ( 2.6149, 5.9736) -- ( 3.9295, 6.5250) node [pos=0.5,below] { 1.2}; \draw [ultra thick] (-0.4602, 2.7379) -- ( 0.8653, 2.1947) node [pos=0.5,below] { 1.2}; \draw [ultra thick] (-0.7957, 3.8720) -- ( 0.1559, 4.5930) node [pos=0.5,below] { 1.2}; \draw [ultra thick] ( 0.1725, 5.9631) -- ( 0.1559, 4.5930) node [pos=0.5,below] { 1.2}; \draw [ultra thick] ( 1.3353, 6.6160) -- ( 2.6149, 5.9736) node [pos=0.5,below] { 1.2}; \draw [ultra thick] ( 3.9295, 6.5250) -- ( 2.6149, 5.9736) node [pos=0.5,below] { 1.2}; \draw [ultra thick] ( 4.9836, 5.7944) -- ( 4.8441, 4.4286) node [pos=0.5,below] { 1.2}; \draw [ultra thick] ( 1.5356, 1.3545) -- ( 0.8653, 2.1947) node [pos=0.5,below] { 1.2}; \draw [ultra thick] ( 2.9057, 1.3064) -- ( 3.7627, 2.0930) node [pos=0.5,below] { 1.2}; \draw [ultra thick] ( 5.1074, 2.5426) -- ( 3.7627, 2.0930) node [pos=0.5,below] { 1.2}; \draw [ultra thick] ( 5.6188, 3.6470) -- ( 4.8441, 4.4286) node [pos=0.5,below] { 1.2}; \draw [ultra thick] ( 1.7034, 2.7712) -- ( 1.3589, 3.9361) node [pos=0.5,below] { 1.2}; \draw [ultra thick] ( 1.3589, 3.9361) -- ( 1.7034, 2.7712) node [pos=0.5,below] { 1.2}; \draw [ultra thick] ( 2.5532, 4.6066) -- ( 1.3589, 3.9361) node [pos=0.5,below] { 1.2}; \draw [ultra thick] ( 3.6359, 3.8562) -- ( 3.1107, 2.7218) node [pos=0.5,below] { 1.2}; \draw [ultra thick] ( 3.1107, 2.7218) -- ( 3.6359, 3.8562) node [pos=0.5,below] { 1.2}; \draw [ultra thick] ( 0.8653, 2.1947) -- (-0.4602, 2.7379) node [pos=0.5,below] { 1.2}; \draw [ultra thick] ( 0.1559, 4.5930) -- ( 0.1725, 5.9631) node [pos=0.5,below] { 1.2}; \draw [ultra thick] ( 3.7627, 2.0930) -- ( 5.1074, 2.5426) node [pos=0.5,below] { 1.2}; \draw [ultra thick] ( 4.8441, 4.4286) -- ( 5.6188, 3.6470) node [pos=0.5,below] { 1.2}; \draw [ultra thick] ( 2.6149, 5.9736) -- ( 1.3353, 6.6160) node [pos=0.5,below] { 1.2}; \draw [ultra thick] (-0.4602, 2.7379) -- (-1.1987, 2.2941) node [pos=0.5,below] { 1.0}; \draw [ultra thick] (-0.7957, 3.8720) -- (-1.7876, 4.2850) node [pos=0.5,below] { 1.0}; \draw [ultra thick] ( 0.1725, 5.9631) -- (-0.7487, 6.5290) node [pos=0.5,below] { 1.0}; \draw [ultra thick] ( 1.3353, 6.6160) -- ( 1.2927, 7.6752) node [pos=0.5,below] { 1.0}; \draw [ultra thick] ( 3.9295, 6.5250) -- ( 4.0765, 7.5776) node [pos=0.5,below] { 1.0}; \draw [ultra thick] ( 4.9836, 5.7944) -- ( 5.9270, 6.2949) node [pos=0.5,below] { 1.0}; \draw [ultra thick] ( 1.5356, 1.3545) -- ( 0.9430, 0.8096) node [pos=0.5,below] { 1.0}; \draw [ultra thick] ( 2.9057, 1.3064) -- ( 3.3483, 0.7252) node [pos=0.5,below] { 1.0}; \draw [ultra thick] ( 5.1074, 2.5426) -- ( 5.7110, 2.0517) node [pos=0.5,below] { 1.0}; \draw [ultra thick] ( 5.6188, 3.6470) -- ( 6.6087, 3.9905) node [pos=0.5,below] { 1.0}; \draw[atomC] ( 1.7034, 2.7712) circle (0.199); \draw[atomC] ( 1.3589, 3.9361) circle (0.168); \draw[atomC] ( 2.5532, 4.6066) circle (0.153); \draw[atomC] ( 3.6359, 3.8562) circle (0.175); \draw[atomC] ( 3.1107, 2.7218) circle (0.204); \draw[atomC] ( 0.8653, 2.1947) circle (0.241); \draw[atomC] ( 0.1559, 4.5930) circle (0.175); \draw[atomC] ( 3.7627, 2.0930) circle (0.250); \draw[atomC] ( 4.8441, 4.4286) circle (0.190); \draw[atomC] ( 2.6149, 5.9736) circle (0.144); \draw[atomC] (-0.4602, 2.7379) circle (0.241); \draw[atomC] (-0.7957, 3.8720) circle (0.210); \draw[atomC] ( 0.1725, 5.9631) circle (0.157); \draw[atomC] ( 1.3353, 6.6160) circle (0.142); \draw[atomC] ( 3.9295, 6.5250) circle (0.151); \draw[atomC] ( 4.9836, 5.7944) circle (0.172); \draw[atomC] ( 1.5356, 1.3545) circle (0.282); \draw[atomC] ( 2.9057, 1.3064) circle (0.287); \draw[atomC] ( 5.1074, 2.5426) circle (0.259); \draw[atomC] ( 5.6188, 3.6470) circle (0.231); \draw[atomH] (-1.1987, 2.2941) circle (0.096); \draw[atomH] (-1.7876, 4.2850) circle (0.077); \draw[atomH] (-0.7487, 6.5290) circle (0.057); \draw[atomH] ( 1.2927, 7.6752) circle (0.047); \draw[atomH] ( 4.0765, 7.5776) circle (0.051); \draw[atomH] ( 5.9270, 6.2949) circle (0.064); \draw[atomH] ( 0.9430, 0.8096) circle (0.112); \draw[atomH] ( 3.3483, 0.7252) circle (0.115); \draw[atomH] ( 5.7110, 2.0517) circle (0.104); \draw[atomH] ( 6.6087, 3.9905) circle (0.087); \end{tikzpicture} \end{center} \end{document}
Теперь пару слов о перспективе. Радиусы атомов (r) вычисляются из z-координат атомов по формуле

Параметр R0 -- это изначальный радиус атома, в качестве него я выбрал ковалентные радиусы углерода (0.7 Å) и водорода (0.25 Å). 0.3 и 0.9 -- просто подгоночные параметры, A -- это разность между максимальным и минимальным значением координат z всех атомов, а ⟨z⟩ -- это среднее значение всех координат атомов, оно нужно чтобы в зависимости от расположения молекулы вдоль оси z у нас не уменьшались/увеличивались все размеры шариков. Естественно, молекула может оказаться плоской, и мы можем расположить её всю в плоскости xy, тогда параметр A окажется равным нулю, поэтому чтобы избежать деления на 0, мы задаём минимально возможное значение этого параметра, равное 0.5 Å.
Полученную последовательность команд мы можем обернуть в простейший Bash-евский скрипт.
#! /bin/bash python3 make_tikz_picture_bond_orders.py pdflatex fig.tex evince fig.pdf &
А вот и получившаяся картинка. В принципе, её можно редактировать как угодно: можно подправить TikZ-овский код, а можно её открыть в каком-нибудь Inkscape и допилить руками. Такое может быть даже проще и веселее.
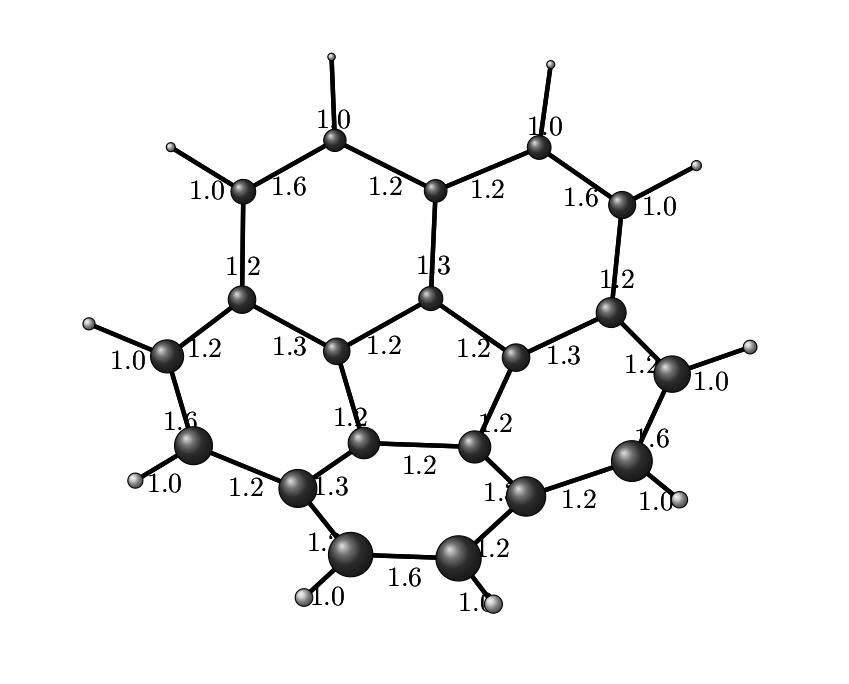
Глава 5. Заключительная.
Конечно, полученный результат всё ещё самопальный отстой, но его можно допилить до ума средствами как самих Python-а/TikZ, так и постфактум, открыв pdf-ку в том же Inkscape. Но суть не в этом, главное, что из говна и палок всегда можно соорудить что-нибудь :) Не уверен, что это достойный вывод, но как-то ничего другого в голову сейчас не лезет.
Ну и естественно, это всё мы можем применить и для куда больших монстров, например, молекул из этой вот статьи, типа C50H20.
mol.xyz
70 energy: -117.275352008181 gnorm: 0.000390187477 xtb: 6.3.3 (5b13467) C -2.68077703943328 -2.91238079355590 0.68845892053073 C 0.27128316214264 0.37286677181363 -2.74314008247056 C 0.60507213164374 1.63822103018138 -2.32805604984469 C 1.14470546196813 -0.70663678298366 -2.43469769398305 C -1.06581084645995 -0.07976153594376 -2.56809812472648 C 1.77225930946232 3.34194992574212 -1.35390644484119 C -1.98283643251999 0.76217241732452 -1.98927003080863 C -1.01875800399212 -1.43900401441490 -2.15147212967076 C -3.55692461220946 -1.82950667221932 0.37905630355287 C -1.89176373398147 -1.86860332754445 -1.18289723879776 C 0.34741614781742 -1.82643527040481 -2.06902919428710 C -3.67216311210816 1.49892949580444 -0.64117931120830 C 0.75242813633749 -2.61847110182269 -1.02333569383271 C 2.29555737015994 -0.45112940559788 -1.73107322169934 C -2.87238808071641 2.62222207085053 -1.00799627676733 C 0.66463461520378 -3.86112902965567 0.89031958102536 C -0.18920704342200 -3.24531710310039 -0.12895583438488 C 2.05676673592769 -2.48502136815445 -0.42298550587013 C 3.95825068512170 -0.66497090027774 -0.18068279856657 C -1.69755571565036 2.15512408221106 -1.74879321174699 C -3.03679366568025 0.27415234921752 -1.13455439255542 C 2.00590199441865 -3.40709107355358 0.71472798937060 C -2.99082756412136 -1.05357171075259 -0.72758318373668 C -1.52370227486734 -2.86686273094032 -0.20948195675439 C -0.39146450717046 2.59725613007549 -1.91977290964893 C 3.91104920579450 0.69851566956800 -0.59860294748331 C 1.90335653160282 1.94647130553701 -1.78127674351813 C 0.40182204522241 3.73059083372130 -1.43661584908383 C 2.83556608650419 -1.39118412041965 -0.78017032846857 C 2.75652657231956 0.89199780089277 -1.47998501347163 H 0.71897816971423 6.94953967009544 -0.48148528448535 C 4.99431155891219 -1.09605306896690 0.62471035753650 C 2.72105457751250 4.26653693910675 -0.96296095556863 C 2.69348079643417 -4.80311075070405 2.53786543979124 C 0.04038665207252 5.02674475351316 -1.12474813797369 C 3.00350369902655 -3.89222211250683 1.53774104921398 H -4.82042850461680 5.07437609405038 0.24363138915910 H -4.49373585157531 -4.36091811325598 3.13578717982119 C 0.37989353033973 -4.78035006216538 1.88121158570339 C -3.03143028976537 -3.81288232917844 1.67539943210881 H 5.07666322596571 -2.12973220473117 0.91419866700697 H 1.16610201764482 -5.96608912900056 3.47724162265726 C 1.39404265134009 -5.24298807813292 2.70798189193439 C -4.22237245502503 -3.65015380568231 2.36912651549759 C -4.74523454500569 -1.69470802282653 1.07018658671297 H -6.00451448165837 -2.49367353365328 2.60227071341764 C -4.86275930627008 1.69930182583409 0.02982253863623 H -0.61535214349515 -5.17261507331524 2.00242406849407 C -3.29834565540937 3.89653803853535 -0.68769832762239 C 4.90198296445000 1.57101884068675 -0.19276970635118 H -2.73557885272309 4.76143849008124 -0.99481217498725 C -4.48985271871798 4.07512095504141 0.00114134487655 H 6.70052705225601 1.80716708714242 0.93899270887624 H 3.08207991140213 6.27938902195787 -0.33886179434377 C 1.00704259699109 5.93589682978381 -0.71842348790248 H -0.97982786218491 5.35578076402524 -1.22462642034273 C -5.26468521546126 2.98685996970108 0.35651988760019 H -5.50538400831336 0.87121774308353 0.27556339882176 H 4.02976647919106 -3.60017823056423 1.39430701088472 H 3.47890531662068 -5.18317486268750 3.17445966634839 H 3.76631271635534 4.00982807793270 -0.93818051057439 C 2.33474085104098 5.55937632045060 -0.63829226138629 H -5.44356464835715 -0.91358199001587 0.82268906061483 C -5.07119650320345 -2.60105093221218 2.06937269377368 H -6.19951205810888 3.13743613055288 0.87615264205200 H 6.78191855437962 -0.54394998060052 1.65962919313397 C 5.92542821547799 1.12146646469345 0.62975329785856 H 4.91319519878343 2.59233680788850 -0.53315466435521 H -2.40926221725034 -4.66382359262515 1.89422285207033 C 5.97115702191632 -0.19949789292965 1.03464030503872
bonds.dat
1 40 1.425 2 3 1.377 3 2 1.377 4 14 1.377 5 7 1.377 6 33 1.425 7 5 1.377 8 10 1.377 9 45 1.425 10 8 1.377 11 13 1.377 12 47 1.425 13 11 1.377 14 4 1.377 15 49 1.425 16 39 1.425 17 24 1.471 18 29 1.471 19 32 1.425 20 25 1.471 21 23 1.471 22 36 1.425 23 21 1.471 24 17 1.471 25 20 1.471 26 50 1.425 27 30 1.471 28 35 1.425 29 18 1.471 30 27 1.471 31 55 0.971 32 19 1.425 33 6 1.425 34 43 1.446 35 28 1.425 36 22 1.425 37 52 0.971 38 44 0.971 39 16 1.425 40 1 1.425 41 32 0.955 42 43 0.971 43 34 1.446 44 64 1.446 45 9 1.425 46 64 0.971 47 12 1.425 48 39 0.955 49 15 1.425 50 26 1.425 51 49 0.955 52 57 1.446 53 67 0.971 54 62 0.971 55 62 1.446 56 35 0.955 57 52 1.446 58 47 0.955 59 36 0.955 60 34 0.971 61 33 0.955 62 55 1.446 63 45 0.955 64 44 1.446 65 57 0.971 66 70 0.971 67 70 1.446 68 50 0.955 69 40 0.955 70 67 1.446 1 9 1.268 2 4 1.162 3 27 1.167 4 2 1.162 5 8 1.162 6 28 1.268 7 21 1.167 8 5 1.162 9 1 1.268 10 24 1.167 11 8 1.162 12 15 1.268 13 17 1.167 14 30 1.167 15 12 1.268 16 22 1.268 17 13 1.167 18 13 1.167 19 26 1.268 20 7 1.167 21 7 1.167 22 16 1.268 23 10 1.167 24 10 1.167 25 3 1.167 26 19 1.268 27 3 1.167 28 6 1.268 29 14 1.167 30 14 1.167 32 70 1.415 33 62 1.415 34 36 1.415 35 55 1.415 36 34 1.415 39 43 1.415 40 44 1.415 43 39 1.415 44 40 1.415 45 64 1.415 47 57 1.415 49 52 1.415 50 67 1.415 52 49 1.415 55 35 1.415 57 47 1.415 62 33 1.415 64 45 1.415 67 50 1.415 70 32 1.415 1 24 1.081 2 5 1.162 3 25 1.167 4 11 1.162 5 2 1.162 6 27 1.081 7 20 1.167 8 11 1.162 9 23 1.081 10 23 1.167 11 4 1.162 12 21 1.081 13 18 1.167 14 29 1.167 15 20 1.081 16 17 1.081 17 16 1.081 18 22 1.081 19 29 1.081 20 15 1.081 21 12 1.081 22 18 1.081 23 9 1.081 24 1 1.081 25 28 1.081 26 30 1.081 27 6 1.081 28 25 1.081 29 19 1.081 30 26 1.081 33 61 0.955 34 60 0.971 35 56 0.955 36 59 0.955 39 48 0.955 40 69 0.955 43 42 0.971 44 38 0.971 45 63 0.955 47 58 0.955 49 51 0.955 50 68 0.955 52 37 0.971 55 31 0.971 57 65 0.971 62 54 0.971 64 46 0.971 67 53 0.971 70 66 0.971