На сегодняшний день применяется не менее четырёх способов классификации вариантов SARS-CoV-2 (или hCoV-19). Проще всего с непривычки запутаться в ветвях филогенетического дерева (кладах), по-разному обозначаемых двумя конкурирующими организациями – GISAID (gisaid.org) и Nextstrain (nextstrain.org). Лавинообразное нарастание количества вариантов вируса усложняет восприятие и понимание подобных обозначений, поэтому деление на клады постепенно вытесняется делением на линии, обозначенным как PANGOLIN (Phylogenetic Assignment of Named Global Outbreak LINeages). Для такого деления используется секвенирование вирусных геномов и построение их полного филогенетического дерева.

https://www.eurosurveillance.org/content/figure/10.2807/1560-7917.ES.2020.25.32.2001410.f1
Кроме того, 31 мая Всемирная организация здравоохранения присвоила ключевым вариантам SARS-CoV-2 простые, легко произносимые и запоминающиеся обозначения с использованием букв греческого алфавита (Альфа, Бета, Гамма, Дельта и т.д.). Некоторые такие варианты имеют и «географические» названия, соответствующие странам их происхождения (Альфа – британский, Бета – южноафриканский, Гамма – бразильский, Дельта – индийский, Лямбда – перуанский, и т.д.), но первоисточники многих вариантов вируса неизвестны, а Бразилия стала прародительницей уже двух вариантов – Гамма и Зета. Поэтому географические названия фигурируют в основном в сообщениях СМИ.
Ещё варианты вируса группируют по степени опасности их распространения. Наиболее опасные обозначают сокращением VOC (Variants of Concern, т.е. вызывающие наибольшую обеспокоенность). Во вторую группу попали «варианты интереса» - VOI (Variants of Interest). Третья группа - это «варианты тревоги» (Alerts for Further Monitoring). Но это по классификации ВОЗ (WHO) и европейского агентства ECDC (European Centre for Disease Prevention and Control). В США (в CDC) последнюю группу обозначают как VOHC (Variants of High Consequence).
Четвёрку наиболее опасных и распространённых вариантов вируса (VOC) образуют Альфа (британский), Бета (южноафриканский), Гамма (бразильский) и Дельта (индийский). Что касается исходного штамма («китайского»), то он в этой классификации не фигурирует, поскольку его геном считается референсным.
")
https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/
Четырём менее распространённым вариантам, образующим группу VOI, также присвоены буквенные (греческие) обозначения (Эта, Йота, Каппа и Лямбда).

Недавно в группу VOI входили ещё варианты Эпсилон (B.1.427/B.1.429), Зета (P.2) и Тета (P.3), но сравнительно слабое распространение позволило переместить их в группу аутсайдеров (VOHC или Alerts for Further Monitoring) и лишить греческих обозначений.
Из-за нестабильности эпидобстановки списки вариантов в этих группах корректируются ежемесячно. И особенно часто – в группе «третьесортных» возбудителей COVID-19 .

Идентифицируют варианты вируса при помощи полногеномного секвенирования, проводимого методами NGS (Next Generation Sequencing). Хотя в России для этих целей иногда пытаются использовать секвенирование по Сэнгеру (капиллярное). Счётчик, показывающий общее количество секвенированных геномов и их распределение по странам, имеется на главной странице сайта CNCB (China National Center for Bioinformation) Китайской академии наук.

К началу августа в базе данных GISAID (с ограниченным доступом) насчитывалось уже больше двух с половиной миллионов записей геномных последовательностей SARS-CoV-2. В базе данных NCBI (с открытым доступом) содержалось 1,057,233 нуклеотидных последовательностей вируса. В том числе 29 полных геномов – из России (к 07.08.2021). На страничке российского проекта VGARus (Virus Genome Aggregator of Russia) 7 августа было указано, что в этой базе данных Роспотребнадзора числится 21623 нуклеотидных последовательностей – 11,498 полных геномов и 10,125 их фрагментов.
Регулярное секвенирование вариантов SARS-CoV-2 стало незаменимым способом мониторинга эпидобстановки во многих странах. В наиболее наглядном виде результаты такого мониторинга представлены на сайте CoVariants (https://covariants.org/), разработанном командой Эммы Ходкрофт из швейцарского Института социальной и превентивной медицины Университета Берна совместно с Институтом биоинформатики (SIB Swiss Insitute of Bioinformatics, Switzerland). На этом сайте можно найти краткие описания свойств наиболее интересных мутаций и вариантов, а также множество интерактивных графиков, демонстрирующих динамику распространения вариантов вируса как в отдельных странах, так и в их регионах. Правда, региональные данные там представлены только для США (по штатам) и для Швейцарии (по кантонам, поделённым на 6 регионов).
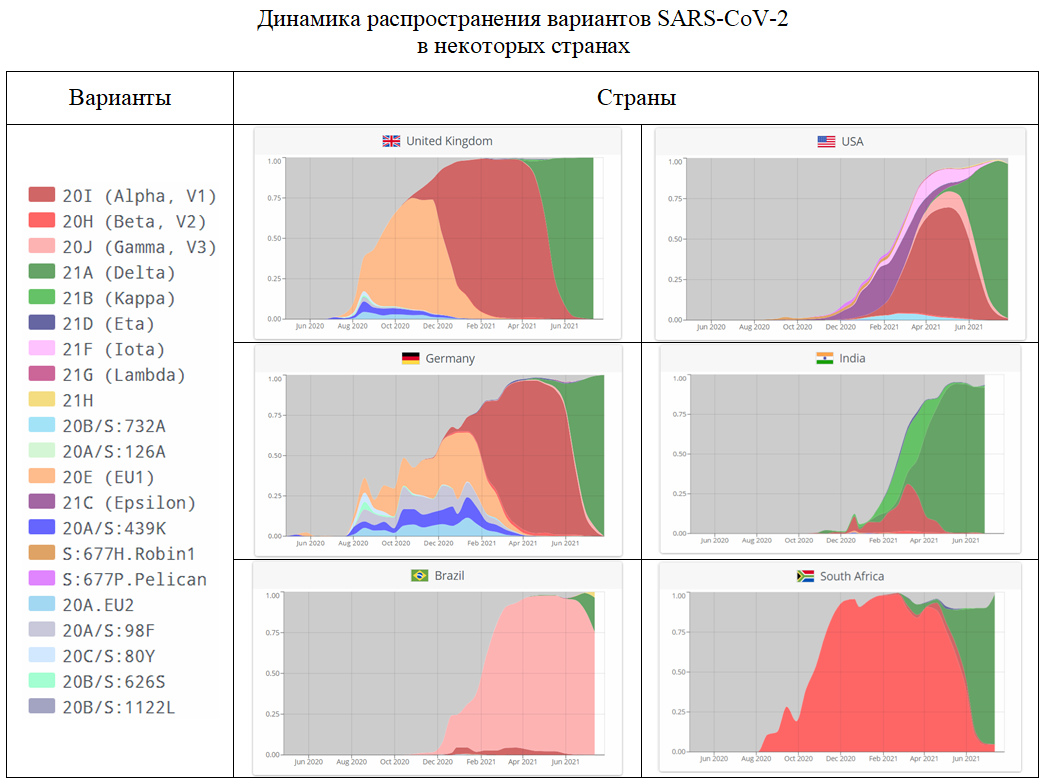
Общая тенденция вполне очевидна – к августу в большинстве стран основным стал вариант Дельта. Даже в ЮАР, в которой совсем недавно господствовал вариант Бета. Исключениями являются некоторые страны Южной и Центральной Америки, в которых основными пока остаются их «родные» южноамериканские варианты (Бразилия - Гамма, Перу – Лямбда, Чили – Гамма + Лямбда). Но, скорее всего, и они скоро будут вытеснены более контагиозным индийским возбудителем COVID-19. Начальную стадию этого процесса можно увидеть на бразильском графике. А завершающую – на графике США, где варианты Гамма и Лямбда ещё пару месяцев назад встречались довольно часто.
В России вариант Дельта стал причиной третьей волны эпидемии COVID-19. В первой половине июля из 244 секвенированных вирусных геномов 242 были идентифицированы как Дельта, один – как Альфа, и только один относился к прочим вариантам, которые преобладали в начале эпидемии.
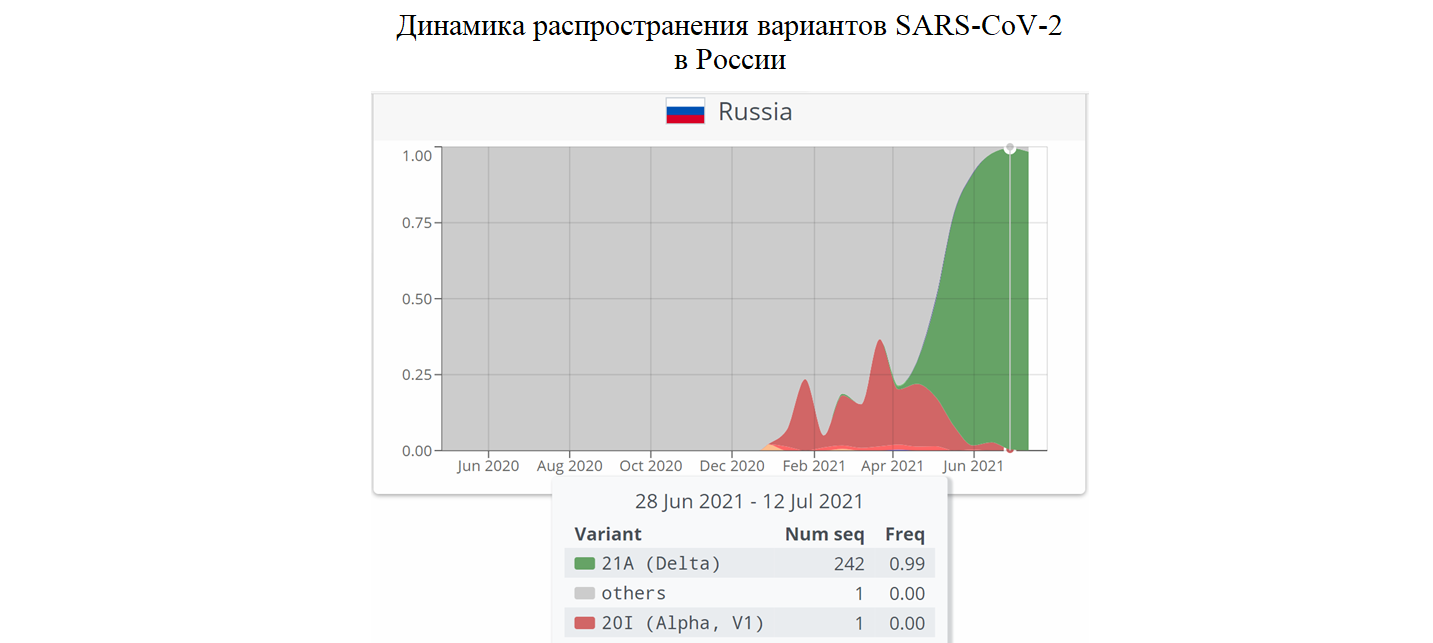
Для построения этого графика швейцарцы использовали данные из GISAID и NCBI. Более подробную картину, по-видимому, может дать отечественная база данных VGARus, но доступ к ней предоставляется только учреждениям Роспотребнадзора. А открытая для просмотра карта распределения вариантов вируса абсолютно неинформативна: https://genome.crie.ru/app/index
Открытый доступ к геномным последовательностям SARS-CoV-2 имеется на сайте Национального центра биотехнологической информации США (NCBI Virus (nih.gov)). Точнее – на странице GenBank, созданной специально для поиска, анализа и скачивания из этой базы данных содержащихся в ней нуклеотидных последовательностей вариантов этого вируса. Меню в левой колонке позволяет определить характер последовательности (полная или неполная), страну её происхождения и линию по классификации PANGO. На правой панели при этом отображается список последних записей в GenBank, в котором можно выбрать конкретную запись и скачать её содержимое в удобном для анализа формате. А если запись хорошо аннотирована, то можно скопировать последовательности отдельных генов вируса. Например, гена S, отвечающего за синтез спайк-белка. Или просто получить идентификационные номера нуклеотидных последовательностей генов и геномов для их последующего анализа.

Располагая такой информацией можно сравнить варианты между собой и найти наиболее вариабельные участки вирусного генома («горячие точки» эволюции), изменение которых улучшает его связывание с рецептором, повышает контагиозность и/или придаёт способность преодолевать иммунитет.
Сравнительный анализ подобного рода проводился неоднократно, причём не только с тысячами нуклеотидных последовательностей вариантов SARS-CoV-2, но и с геномами его ближайших родственников (первого SARS, ближневосточного MERS и некоторых коронавирусов летучих мышей). И уже известно, что чаще всего мутации происходят в гене S, кодирующем спайк-белок, доменная структура которого описана во многих статьях. Рисунок с координатами его доменов (и субдоменов) можно найти, например, здесь.

При наличии нуклеотидных последовательностей разных вариантов вируса можно посмотреть локализацию мутаций в гене S. Например, при помощи программы NUCLON.

На этом графике видно, что мутации сконцентрированы в основном в трёх зонах - в NTD (N-terminal domain), в RBD (Receptor binding domain) и в районе фуринового сайта расщепления белка (680-685), что может указывать на ускоренную эволюцию этих участков вирусного генома. И на эпитопы белка S, антитела к которым обладают иммунопротективными свойствами. Хотя появление некоторых мутаций может быть обусловлено и другими причинами.
Для надёжного определения причин появления мутаций и более точного определения границ иммунопротективных эпитопов имеющейся информации ещё недостаточно. Но это замечательно, поскольку свидетельствует о высокой консервативности вирусного генома. И о том, что борьба с ним ещё не проиграна.
Если «присмотреться» к двум вариантам, которые внесли наибольший вклад в две последние волны COVID-19 в России (Альфа и Дельта), то можно обнаружить в спектре их мутаций некоторые интересные особенности.

У варианта Альфа в богатом иммунопротективными эпитопами N-терминальном домене (NTD) находятся две небольшие делеции (H69-V70, Y144). У варианта Дельта на этом участке белка находятся три аминокислотные замены и одна делеция (T19R, K77T, G142D, E156-F157-R158). В рецептор-связывающем домене (RBD) каждый вариант имеет по две мутации (Альфа – N501Y и A570D; Дельта – L452R и T478K). Причем одинаковые мутации отсутствуют, и только две пары мутаций расположены достаточно близко, чтобы принадлежать к общим эпитопам. Одну пару образуют делеция H69-V70 (Альфа) и замена K77T (Дельта). Вторую – делеция Y144 (Альфа) и замена G142D (Дельта).
Отсюда следует, что по отдельности мутации в вариантах Альфа и Дельта не могут перекрыть все иммунопротективные эпитопы. И что вакцины на основе S-белка из референсного варианта вируса (Спутник V и КовиВак) должны защищать от этих вариантов, хотя и с меньшей эффективностью.
Подобный анализ мутаций позволяет сделать ещё один вывод - естественный иммунитет у переболевших вариантом Альфа будет защищать от заражения вариантом Дельта хуже, чем вакцинация референсным вариантом белка S.
Эти выводы не более чем подтверждают уже известные истины, но получены не эпидемиологами и паталогоанатомами в результате многочисленных наблюдений за пациентами, а простым анализом вариабельности вирусного белка S.
11.08.2021 г.
