 В современном мире анализа данных использовать только один метод или только один подход означает, что рано или поздно ты столкнешься с фактом, как сильно ты ошибался. Для анализа данных комбинируют различные методики, сравнивают результат и на основании сравнения уже делают более точные прогнозы. В программе ZINBA использован именно такой подход. Разработчики объединили разнообразные методы анализа DNA-seq экспериментов в едином пакете. Этот пакет написан для программы статистической обработки данных R. Что же делает ZINBA? Находит различные обогащенные регионы даже в тех случаях, когда некоторые из них были усилены, например, химически или имеют разную степень соотношения сигнал-шум.
В современном мире анализа данных использовать только один метод или только один подход означает, что рано или поздно ты столкнешься с фактом, как сильно ты ошибался. Для анализа данных комбинируют различные методики, сравнивают результат и на основании сравнения уже делают более точные прогнозы. В программе ZINBA использован именно такой подход. Разработчики объединили разнообразные методы анализа DNA-seq экспериментов в едином пакете. Этот пакет написан для программы статистической обработки данных R. Что же делает ZINBA? Находит различные обогащенные регионы даже в тех случаях, когда некоторые из них были усилены, например, химически или имеют разную степень соотношения сигнал-шум.Сначала я рассчитывал сделать обзорную статью, о программном продукте ZINBA, но чем больше читал об использованных в ней методах, тем глубже закапывался в алгоритмах и определениях. И когда в дальнейшем конспектировал полученные знания, осознал тот факт, что данных набралось уже не на одну статью и без вступления трудно будет прикоснуться к сути вопроса. В этом топике я привожу краткие выдержки из статей, упомянутых в описании программы ZINBA, дополняя их собственными замечаниями. Буду ждать ваши комментарии, чтобы вместе докопаться до истины.
Далеко идущие планы регуляторной биологии заключаются в том, чтобы познать, каким образом геном кодирует разнообразие механизмов генной экспрессии. Связь между кодированием и этими механизмами обнаруживается благодаря возможности нахождения сайтов связывания белков по всему геному с помощью иммунопреципитации хроматина (ChIP) и экспрессии генов (RNA-seq). Ходить за доказательствами далеко не надо, есть проект Encyclopedia of DNA Elements (ENCODE [1]), в рамках которого распознается большинство функциональных элементов генома. Первые шаги делались с помощью микрочипового (“microarray”) способа, но глубокое секвенирование ДНК (ChIP-seq & DNA-seq, иногда встречается термин NGS next generation sequencing) догнало и обогнало эту технологию.
RNA-seq и ChIP-seq обладают следующими положительными отличиями: увеличивают точность, специфичность, чувствительность измерений и позволяют рассматривать сразу весь геном. В общем-то RNA-seq и ChIP-seq имеют некоторую схожесть с таким предшественником, как микрочип, но частности сильно разнятся. Но секвинирование все ещё страдает рядом трудностей, хотя и сильно лучьше чем микрочип в этих вопросах. В связи с тем, что нарезаемые фрагменты ДНК довольно коротки, есть вероятность, что фрагменты могут несколько раз встречаться в геноме, быть участками повторяемой (repetitive) части ДНК или же оказаться общими фрагментами для некоторых старт сайтов.
Считается, что обработка данных должна рассматривать ChIP реакцию, как обогащение. Потому что примерно 60-99% фрагментов в подготовленном растворе ДНК обычно является фоном, а оставшиеся 1-40% относятся к фрагментам, белок которых смог связаться (cross-link) с антителом [2]. Именно эта наименьшая часть раствора будет секвенирована. Тот факт, что уровень загрязнения высок, должен учитываться при вычислении обогащения. Количество ридов на геном в некоторых случаях несравнимо мало, например, для генома млекопитающего количество считанных ридов меньше, чем 1% от общей длины генома. Данные получаемые при секвенировании требуют, очевидно, новых алгоритмов и программного обеспечения.
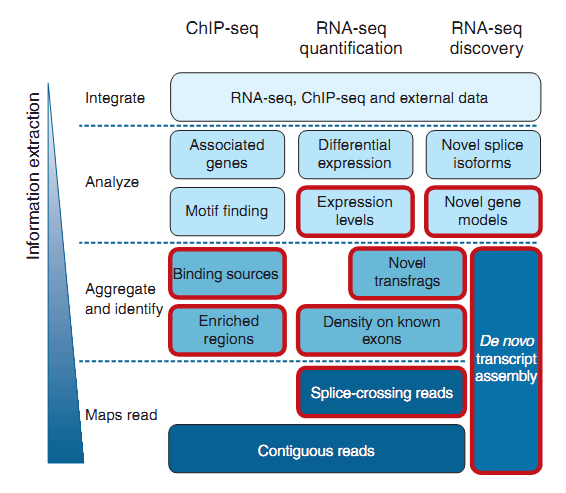 Рис. 1
Рис. 1Этот слайд схематично отображает иерархию ChIP-seq и RNA-seq анализа. Обычно анализ проходит все этапы снизу вверх. Разные программы применяются на каждом уровне схемы и иногда разделяются на программы специально для ChIP-seq и RNA-seq. Выходные данные одной программы являются входом для другой (pipeline). Как видно из схемы, все программы сначала проходят через определение ридов на геноме(Maps read), за исключением сборки транскриптов, результатом которой являются предполагаемые транскриптомы[3]. После нахождения ридов на геноме, выход такой программы передается на вход программы сбора и идентификации данных (Aggregate and identity). Например, для определения обогащенных регионов или плотности ридов на известной аннотации. Также эти программы могут в себя включать некоторый последующий анализ как сайтов привязывания (для DNA-seq), так и новых неизученных фрагментов транскрипции-изоформизма (для ChIP-seq). Далее проводится анализ более высокого уровня. Он включает в себя определение консервативных мотивов или уровня экспрессии, результатом которых может стать открытие новой модели гена. Уровень интегрирования (Integrate) показывает, что для текущей обработки данных могут использоваться ранее полученные результаты (необязательно в рамках текущего эксперимента, лаборатории или страны).
Так что же мы получаем после того, как риды были найдены на геноме? Как выглядят эти данные и чем отличаются друг от друга? На ДНК есть участки (binding sites), к которым может прикрепиться тот или иной белок, эти участки мы будем называть сайтами связывания или просто сайтами. Например, CTCF сайт означает, что белок CTCF может закрепиться на этом участке. Ученые нашли антитело к CTCF белку и с помощью антитела смогли осадить соответствующие участки ДНК. Но, как мы уже знаем, вместе со специальными участками к антителу могут прикрепляться и произвольные. Остальные методы работают аналогично: белок закрепляется на ДНК, антитело закрепляется на белке, только выборка участков ДНК меняется. Таким образом, специфика выбираемых участков накладывает свой отпечаток, и ландшафт рисунка у каждого метода будет получаться свой: где-то узкие пики, как в CTCF методе, график a; где-то крутые пики с большой прилегающей территорией, например RNA polymerase II метод, график b; при осаждении хистонной модификации получается сплошная «бахрома”, это связано с особеностью расположения хистонов, рисунок c. На графиках по оси „x“ откладывается координата на хромосоме, а по оси „y“ плотность ридов. Данные для рисунка взяты из работы [4].
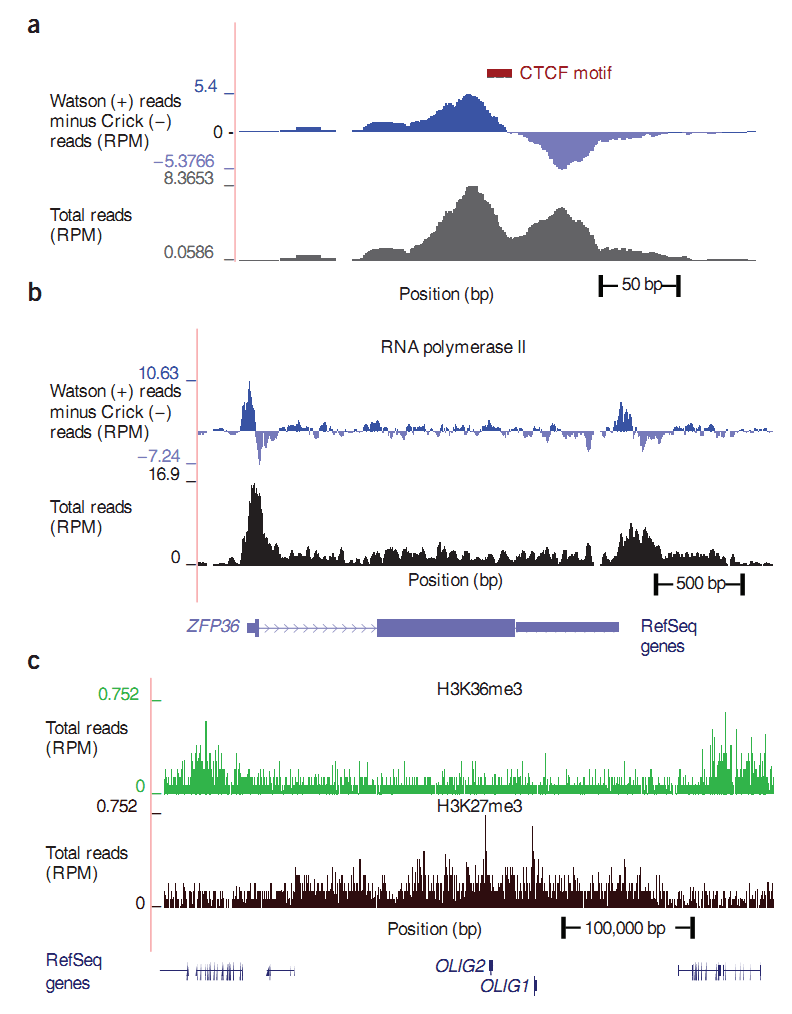 Рис. 2
Рис. 2Наличие фона вынуждает алгоритмы пытаться оценить его эмпирическим путем на основании “контроля”. Контроль — это дополнительный эксперимент, проводимый с исходным раствором со специальными (preimmune) антителами, с которыми ничего не должно соединяться, но все-таки случайным образом соединяется. В результате этого соединения получается некоторый произвольный фон. Некоторые алгоритмы моделируют возможный фон, основываясь только на полученных данных, без контроля. Какой бы из подходов ни был выбран (с контролем и без контроля), распределение фоновых ридов до конца унифицировать нельзя, так как уровень фона зависит, как от типа клеток и ткани, из которой они получены, так и от способа осаждения и т.д. Применяемая технология для усиления (amplification, PCR) и секвенирования также может добавить искусственных обогащений (artifacts) или усилить один участок больше, чем другой. Существуют алгоритмы, которые позволяют находить обогащенные участки для каждого эксперимента. Хотя каждый из алгоритмов заточен под соответствующие эксперименту данные, предположение, лежащее в основе алгоритма, не всегда подходит возможному множеству обогащенных участков, найденных при помощи DNA-seq [2].
Ниже, для сравнения данных, приведены графики из более новой статьи по ZIMBA. Схожие графики смазываются из-за различия в масштабах между первым и вторым рисунками, но выглядят соответствующе вышеприведенному разъяснению про технологию DNA-seq (см. Рис. 2).
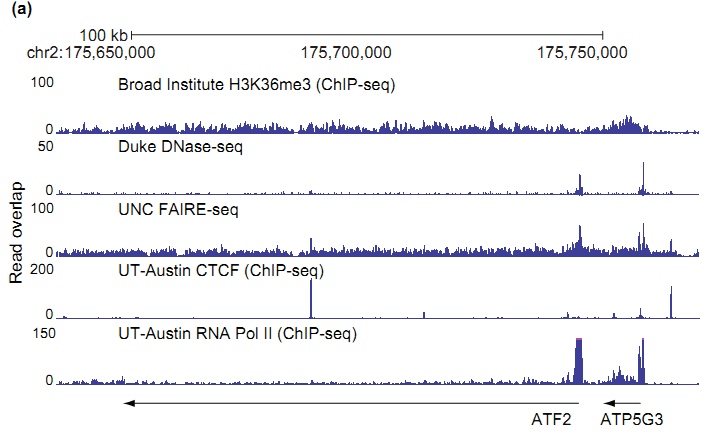
Рис. 3
На рисунке 3 дополнительно к предыдущим графикам отображены ландшафты следующих DNA-seq экспериментов: ДНКазо чувствительная выборка (DNase-seq)[5] и изоляция/фиксация белков с помощью формальдегида (FAIRE-seq)[6].
ДНК — это двойная спираль. Эта фраза сама по себе популярна, но очень часто её забывают. Что она означает в нашем случае? А то, что программа, которая будет искать риды на геноме, выдаст вместе с координатами на хромосоме и то, с какой «стороны» спирали (strand) этот рид находится. Спирали разделяют на положительную (sense) и неположительную (nonsense/antisense). Механизм получения ридов таков, что большинство ридов будет располагаться с двух разных сторон относительно белка, связанного с ДНК. Причем в большинстве случаев, риды будут находиться с 5’ конца соответствующей спирали. Поэтому риды с двух сторон будут стремиться к центру сайта связывания белка. Рисунок 4 это схематично отображает, красные точки — это риды со стороны 5’ конца, а в центре находится белок, связанный с ДНК.
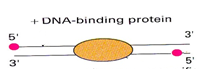
Рис. 4
Таким образом, для нахождения центра сайта связывания белка с ДНК построим следующий график. Для этого по оси “y” откладываем разницу между количеством ридов с положительной и отрицательной сторон, по оси “x” откладываем координату рида, соответствующую хромосоме. На рисунке 5 риды с положительной стороны отображены красным, а с отрицательной — синим. Получившийся график пересекает ноль очень близко к центру сайта связывания белка (см. нижний график Рис. 5). Существуют алгоритмы, которые с точностью до нуклеотдида определяют сайты связывания.
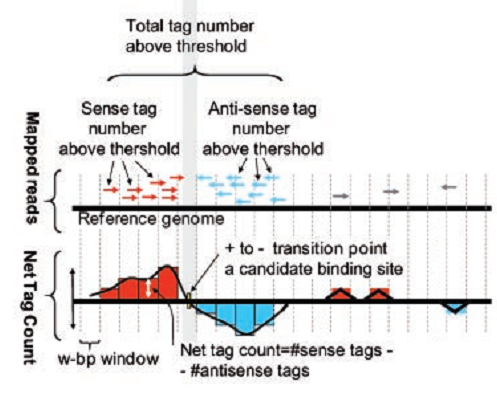
Рис. 5
В этом топике я постарался кратко ознакомить с материалом, являющимся объектом исследования. Были показаны отличия ландшафтов соответствующих DNA-seq экспериментов, объяснен способ приблизительного нахождения центра сайта связывания белка. Впереди огромная работа: предстоит понять отличия алгоритмов в зависимости от ландшафта, научиться находить пики и постараться отличать их от искусственных, выяснить, какие регионы генома, лежащие в окрестностях пика, можно считать обогащенными, и все это для того, чтобы начать рассуждение о работе ZINBA.
1. Birney, E., et al., Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature, 2007. 447(7146): p. 799-816.
2. Pepke, S., B. Wold, and A. Mortazavi, Computation for ChIP-seq and RNA-seq studies. Nat Methods, 2009. 6(11 Suppl): p. S22-32.
3. Barski, A. and K. Zhao, Genomic location analysis by ChIP-Seq. J Cell Biochem, 2009. 107(1): p. 11-8.
4. Barski, A., et al., High-resolution profiling of histone methylations in the human genome. Cell, 2007. 129(4): p. 823-37.
5. Boyle, A.P. and T.S. Furey, High-resolution mapping studies of chromatin and gene regulatory elements. Epigenomics, 2009. 1(2): p. 319-329.
6. Giresi, P.G. and J.D. Lieb, Isolation of active regulatory elements from eukaryotic chromatin using FAIRE (Formaldehyde Assisted Isolation of Regulatory Elements). Methods, 2009. 48(3): p. 233-9.
Review is prepared by Andrey Kartashov, Cincinnati, OH, porter@porter.st
corrected by Ekaterina Morozova, ekaterina@porter.st.
