 Из всех известных мне технических и естественных наук, пожалуй, именно о биоинформатике представление у людей самое плохое. Оно либо в той или иной степени неверное, либо его нет совсем. Когда два года назад я начал заниматься бионформатикой, знаний об этой науке у меня самого не было ровным счетом никаких. Со временем я лучше стал представлять, какие задачи стоят перед биоинформатиками, чем они пользуются, и что может являться результатом их работы.
Из всех известных мне технических и естественных наук, пожалуй, именно о биоинформатике представление у людей самое плохое. Оно либо в той или иной степени неверное, либо его нет совсем. Когда два года назад я начал заниматься бионформатикой, знаний об этой науке у меня самого не было ровным счетом никаких. Со временем я лучше стал представлять, какие задачи стоят перед биоинформатиками, чем они пользуются, и что может являться результатом их работы.У биоинформатиков нет никаких пробирок, реагентов, бактерий, белых халатов. Главные инструменты у них – ноутбук, ручка с бумагой или белая доска с маркером – в общем, всё как у программистов. Да и сама работа очень сильно похожа на работу в IT компании, а лаборатория – на небольшой отдел разработки. А в чем же тогда отличия? Что ж, попробую ответить.
Во-первых, задачи в основном алгоритмические. То есть перед тем как написать программу, надо прочитать несколько статей, подумать самому, обсудить свои идеи с коллегами и только потом приступать к реализации. Во-вторых, работать приходится с большими объемами данных, а поэтому реализация должна быть максимально эффективной. Однако даже эффективная, логичная и идеально отлаженная программа может не дать желаемый результат. Основная причина тому – биологическое происхождение данных, а значит огромное количество ошибок и существенное различие между данными от разных лабораторий.
Еще одно, пожалуй, самое видимое отличие биоинформатики от программирования – это исследования и публикации. Биоинформатика – это наука, а значит просто необходимо быть в курсе всего, что происходит в мире. Для этого и существуют многочисленные конференции, сотрудничества с лабораториями из других стран и, безусловно, публикации – о своих достижениях тоже необходимо рассказывать всем. Без всего этого можно усердно изобретать велосипед.
В общем и целом, впечатление о биоинформатике именно такое, но лучше всего рассказать это на примере, тем более что такой есть, и совсем недалеко. Но обо всем по порядку.
Лаборатория алгоритмической биологии
В 2010-м году в России была запущена программа «мегагрантов». Под руководством ведущих западных ученых (в большинстве случаев давно уехавших из России) стали создаваться новые научные лаборатории. Одной из таких стала лаборатория алгоритмической биологии при СПбАУ под руководством Павла Певзнера – одного из самых известных ученых в своей области. Павел закончил МФТИ, но достаточно скоро после этого уехал в США, занялся Computer Science (а если быть точным, именно биоинформатикой) и сейчас является профессором Университета Калифорнии в Сан-Диего.
Перед тем как рассказать, чем же именно занимаются в лаборатории, стоит ввести читателей в курс дела.
Немного о геномике
Уверен, что каждый читатель слышал слово геном. Для биологов геном – это молекулы ДНК – длинные цепочки, состоящие из четырех нуклеотидов, организованные в хромосомы, свёрнутые в ядре клетки. Мы же видим геном как строку, состоящую опять же из четырех символов (A, C, G, T). Длина генома может достигать миллиардов или даже десятков миллиардов символов. Биологи не умеют считывать геном целиком – только маленькими фрагментами до 150 «букв», да и то с ошибками. Наша задача – восстановить исходный геном по этим кусочкам, или как чаще говорят – собрать.
Для наглядности можно привести такое сравнение: представьте пачку одинаковых газет. Теперь представьте, что эту пачку взрывают и мелкие кусочки бумаги разлетаются, перемешиваются, портятся или даже сгорают целиком. А дальше по куче этого мусора хочется склеить исходную газету.
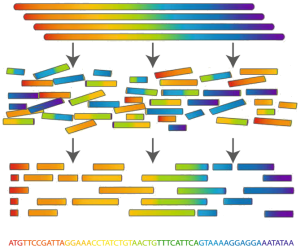 Так же и с геномом. Первые технологии позволяли считывать кусочки генома длиной до нескольких тысяч символов. Эти технологии были невероятно дорогие – на сборку первого человеческого генома были потрачено несколько миллиардов долларов и несколько лет усердной работы сотен сотрудников лабораторий по всему миру. Современные технологии позволяют читать более короткие фрагменты, но на порядок дешевле и в огромном количестве. Обработка гигабайтов входных данных, естественно, производится автоматически. Для этого разрабатываются программы, которые называют геномными сборщиками, или чаще – ассемблерами (от английского assemble). В силу некоторых особенностей исходных геномов (например, повторяющихся регионов), а также большого числа ошибок во входных данных, результатом работы сборщика является не целый геном, а лишь достаточно продолжительные его участки. Чем длиннее полученные участки, чем больше они похожи на исходный геном, тем качественней считается результат.
Так же и с геномом. Первые технологии позволяли считывать кусочки генома длиной до нескольких тысяч символов. Эти технологии были невероятно дорогие – на сборку первого человеческого генома были потрачено несколько миллиардов долларов и несколько лет усердной работы сотен сотрудников лабораторий по всему миру. Современные технологии позволяют читать более короткие фрагменты, но на порядок дешевле и в огромном количестве. Обработка гигабайтов входных данных, естественно, производится автоматически. Для этого разрабатываются программы, которые называют геномными сборщиками, или чаще – ассемблерами (от английского assemble). В силу некоторых особенностей исходных геномов (например, повторяющихся регионов), а также большого числа ошибок во входных данных, результатом работы сборщика является не целый геном, а лишь достаточно продолжительные его участки. Чем длиннее полученные участки, чем больше они похожи на исходный геном, тем качественней считается результат.Задача сборки генома
Если взять задачу сборки генома в самом общем случае – это будет не что иное, как задача о надстроке (shortest superstring problem), которая формулируется следующим образом: найти кратчайшую строку, такую, что каждая строка из заданного набора являлась бы её подстрокой. Эта задача является NP-полной. Но если предположить, что у нас есть все возможные подстроки исходной строки одинаковой длины, задачу можно решить за полиномиальное время. Сборка генома – это именно такой случай. В 2001 году Павлом Певзнером был предложен эффективный подход сборки геномов с использованием графа де Брюйна. Основная идея этого подхода используется почти в каждом современном геномном ассемблере. Однако на практике все сильно усложняется вышеупомянутыми биологическими ошибками, и поэтому основная задача – разработка эвристик для разного рода подзадач, возникающих при сборке геномов.
В лаборатории алгоритмической биологии было решено сфокусироваться именно на разработке ассемблера. Безусловно, на момент создания лаборатории существовало огромное количество геномных сборщиков. Зачем было тогда создавать еще один? На самом деле, задача сборки оказывается намного более широкой, чем кажется на первый взгляд. Биологи производят огромное количество различных типов входных данных, для каждого из которых требуется разработка новых методов, учитывающих их специфику. Кроме того, сборка генома включает в себя большое число этапов и алгоритмов, поэтому даже несмотря на то, что все современные ассемблеры используют один и тот же подход, их результаты могут очень сильно отличаться. Перед лабораторией ставилась задача получить ассемблер, который по многим параметрам превосходил бы существующие.
Путь в биоинформатику
В биоинформатику я попал, можно сказать, случайно. Я учился в магистратуре СПбАУ и, как и каждому студенту, в начале семестра мне необходимо было выбрать научно-исследовательскую работу. Чтобы попробовать себя в новой области, я выбрал биоинформатический проект. По началу пугало то, что, возможно, придется учить биологию, вместо того чтобы разрабатывать и реализовывать алгоритмы. Однако опасения, к счастью, не оправдались – погружение в эту предметную область происходит точно так же, как и в любую другую. Постепенно начинаешь понимать больше, узнаешь что-то новое, и даже если биология была далеко не самым любимым предметом в школе, интерес к ней появляется достаточно быстро. Почти сразу я понял, что биоинформатика – это именно то, чем я хотел заниматься – программирование с элементами исследовательской работы и интересной предметной областью.
Пока я занимался своим проектом, организовалась лаборатория алгоритмической биологии, о которой я упоминал. Летом 2011 я успешно прошел в ней стажировку и остался как постоянный научный сотрудник. Если говорить о лаборатории в целом – огромное количество различных интересных проектов, которые далеко не ограничиваются сборкой геномов, сотрудничество с западными лабораториями, научные конференции, постоянная возможность узнавать что-нибудь новое и, конечно, очень хороший коллектив.
Можно было бы наверное еще долго говорить и о работе в лаборатории, и о биоинформатике в целом, в которой еще море открытых проблем, и о конкретных подходах и алгоритмах в разных задачах. Но нельзя объять необъятное, а посему рассказ будет о чем-то одном и уже в следующий раз. А о чем именно – зависит от ваших пожеланий.
Ссылки
- P. E. C. Compeau and P. A. Pevzner. Genome Reconstruction: A Puzzle with a Billion Pieces (oбзорная статья о сборке геномов)
- P.A. Pevzner, H. Tang, and M. Waterman. An Eulerian path approach to DNA fragment assembly. Proceedings of the National Academy of Science of the United States of America, 98:9748-9753, 2001
