 C тех пор, как я опубликовал первую обзорную статью о биоинформатике, прошел уже почти год. Хотелось бы писать чаще, но как раз биоинформатика отвлекаться и не дает. В предыдущей статье была упомянута задача сборки генома, а также Лаборатория алгоритмической биологии (СПбАУ РАН), в которой ей занимаются. Как уже было сказано, над этой задачей работают во многих университетах мира давно и достаточно успешно. Однако для сборки генома биологи получают огромное количество различных типов данных, каждый из которых имеет свои особенности. Около пяти лет назад появилась технология MDA, открывшая большие возможности в области изучения бактерий. В результате появился тип данных, для которого потребовался новый геномный сборщик. Через несколько лет он был разработан, и не в Стэнфорде или Массачусетсе, а в Санкт-Петербурге, в молодом Академическом университете. Но обо всем по порядку.
C тех пор, как я опубликовал первую обзорную статью о биоинформатике, прошел уже почти год. Хотелось бы писать чаще, но как раз биоинформатика отвлекаться и не дает. В предыдущей статье была упомянута задача сборки генома, а также Лаборатория алгоритмической биологии (СПбАУ РАН), в которой ей занимаются. Как уже было сказано, над этой задачей работают во многих университетах мира давно и достаточно успешно. Однако для сборки генома биологи получают огромное количество различных типов данных, каждый из которых имеет свои особенности. Около пяти лет назад появилась технология MDA, открывшая большие возможности в области изучения бактерий. В результате появился тип данных, для которого потребовался новый геномный сборщик. Через несколько лет он был разработан, и не в Стэнфорде или Массачусетсе, а в Санкт-Петербурге, в молодом Академическом университете. Но обо всем по порядку. Снова о сборке генома
Вкратце напомню базовые понятия. Геном для биологов — это длинные молекулы ДНК, представляющие из себя двойную цепочку из четырех типов нуклеотидов (аденин, цитозин, гуанин и тимин). К примеру, длина цепочки ДНК у бактерии измеряется миллионами нуклеотидов, в то время как длина человеческой ДНК примерно равна 3 миллиардам. Для биоинформатиков геном — это просто большая строка над алфавитом из четырех символов {A, C, G, T}. “Прочесть” всю молекулу ДНК целиком невозможно. Современные технологии позволяют считывать только маленькие кусочки длиной в несколько сотен нуклеотидов из случайных мест, да и то с ошибками. Эти кусочки называются чтениями или ридами (reads), а процесс считывания — секвенированием (sequencing). За один эксперимент производится несколько десятков или даже сотен миллионов чтений. Дальше в дело вступают биоинформатики, разрабатывающие геномные ассемблеры — программы, пытающиеся восстановить исходную последовательность по этим чтениям. Как правило, ассемблерам не удается восстановить весь геном целиком, но из миллионов коротких чтений они позволяют получить десятки последовательностей длиной в сотни тысяч нуклеотидов. Такие последовательности уже могут дальше изучаться биологами.
 Кому же может понадобится собирать и анализировать геном? К примеру, сборка человеческого генома может помочь определить наличие в организме раковых клеток еще на ранних этапах. Еще одна цель — переход к так называемой персональной медицине. Если у каждого человека известен геном, то лекарства и лечение могут прописываться ему не только по симптомам, но еще и с учетом его генетических особенностей. Персональная медицина еще не получила широкое распространение, но некоторые клиники уже предоставляют услуги по секвенированию геномов пациентов. Цена чтения генома постоянно падает, а новые алгоритмы сборки позволяют осуществлять ее быстрее. Тем не менее, одного лишь генома человека недостаточно для полноценной диагностики. В каждом из нас обитает несколько тысяч видов различных бактерий, которые участвуют в огромном количестве внутренних процессов и являются необходимыми для жизни. Сборка геномов этих бактерий (суммарная длина которых превышает длину генома самого человека) позволила бы намного лучше изучить человеческий организм. Однако в их секвенировании есть сложности.
Кому же может понадобится собирать и анализировать геном? К примеру, сборка человеческого генома может помочь определить наличие в организме раковых клеток еще на ранних этапах. Еще одна цель — переход к так называемой персональной медицине. Если у каждого человека известен геном, то лекарства и лечение могут прописываться ему не только по симптомам, но еще и с учетом его генетических особенностей. Персональная медицина еще не получила широкое распространение, но некоторые клиники уже предоставляют услуги по секвенированию геномов пациентов. Цена чтения генома постоянно падает, а новые алгоритмы сборки позволяют осуществлять ее быстрее. Тем не менее, одного лишь генома человека недостаточно для полноценной диагностики. В каждом из нас обитает несколько тысяч видов различных бактерий, которые участвуют в огромном количестве внутренних процессов и являются необходимыми для жизни. Сборка геномов этих бактерий (суммарная длина которых превышает длину генома самого человека) позволила бы намного лучше изучить человеческий организм. Однако в их секвенировании есть сложности. От человека к бактериям
Чтобы секвенировать геном, необходимо иметь достаточно большое количество генетического материала — одинаковых молекул ДНК. Для стандартного эксперимента требуется несколько десятков или сотен миллионов идентичных клеток. В случае многоклеточных организмов, в том числе и человека, это проблемой не является, так как достаточное количество генетического материала содержится в нескольких граммах слюны. Получить столько же бактериальных клеток сложнее — их необходимо выращивать в лабораторных условиях. Проблема заключается в том, что далеко не все бактерии можно культивировать. Например, бактерии, находящиеся внутри кишечника человека, живут в больших колониях и не способны существовать и размножаться отдельно друг от друга. Культивировать одну взятую из колонии бактерию невозможно.
На сегодняшний день существует два принципиально различных подхода к секвенированию некультивируемых бактерий в колониях. Первый, достаточно простой с биотехнологической точки зрения — метагеномика. Он заключается в следующем: берем колонию, выделяем из всех бактерий молекулы ДНК, помещаем их в секвенатор и считываем. В результате мы одновременно получим чтения геномов нескольких десятков различных бактерий — от каких-то больше, от каких-то меньше. У близкородственных бактерий геномы похожи друг на друга, у бактерий разных типов — сильно отличаются, но могут содержать идентичные участки. Эти факторы сильно усложняют сборку такого рода данных. Несмотря на ряд проведенных и проводимых сегодня метагеномных исследований, не могу сказать, что существует хотя бы один ассемблер, который на таких данных давал бы результаты, удовлетворяющие биологов. Как правило, сборщики выдают короткие последовательности, которые содержат большое число ошибок и плохо поддаются анализу. Иногда в метагеномных проектах обнаружение нескольких генов для всего набора различных бактерий уже является успехом.
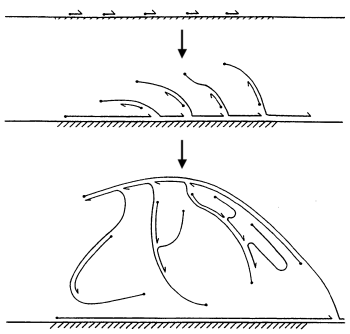 Другой подход секвенирования некультивируемых бактерий был открыт всего несколько лет назад и заключается в достаточно сложном биотехнологическом процессе. Берем одну интересующую нас бактерию из колонии, извлекаем из неё ДНК и сажаем на ДНК несколько “копировальных машин”, которые начинают “бегать” по молекуле и копировать её участки случайным образом. При копировании от молекулы ответвляются новые нити ДНК, на которые в свою очередь тоже может сесть “копировальная машина” и продолжить наращивание генетического материала. В результате мы получаем идентичные копии участков интересующей нас ДНК, которые помещаем в секвенатор. Этот метод секвенирования генома единичной клетки называется Multiple Displacement Amplification (MDA). Казалось бы, все замечательно. Но, увы, и тут есть проблемы. “Копировальные машины” работают сами по себе и управлять ими невозможно. Поэтому какие-то участки ДНК могут быть скопированы десять тысяч раз, а другие — ни разу. В итоге, некоторые регионы будут отсутствовать в сборке попросту из-за нехватки материала. В среднем в результате такого метода можно восстановить 95-99% генома в зависимости от его структуры (некоторые поддаются копированию хуже) и качества эксперимента.
Другой подход секвенирования некультивируемых бактерий был открыт всего несколько лет назад и заключается в достаточно сложном биотехнологическом процессе. Берем одну интересующую нас бактерию из колонии, извлекаем из неё ДНК и сажаем на ДНК несколько “копировальных машин”, которые начинают “бегать” по молекуле и копировать её участки случайным образом. При копировании от молекулы ответвляются новые нити ДНК, на которые в свою очередь тоже может сесть “копировальная машина” и продолжить наращивание генетического материала. В результате мы получаем идентичные копии участков интересующей нас ДНК, которые помещаем в секвенатор. Этот метод секвенирования генома единичной клетки называется Multiple Displacement Amplification (MDA). Казалось бы, все замечательно. Но, увы, и тут есть проблемы. “Копировальные машины” работают сами по себе и управлять ими невозможно. Поэтому какие-то участки ДНК могут быть скопированы десять тысяч раз, а другие — ни разу. В итоге, некоторые регионы будут отсутствовать в сборке попросту из-за нехватки материала. В среднем в результате такого метода можно восстановить 95-99% генома в зависимости от его структуры (некоторые поддаются копированию хуже) и качества эксперимента.Проблемы технологии MDA
Количество раз, которое нуклеотид в геноме считывается в процессе секвенирования, называется его покрытием. При стандартном секвенировании покрытие всех нуклеотидов примерно одинаковое (пример графика покрытия на левом рисунке снизу). При использовании технологии MDA мы получаем данные с крайне неравномерным покрытием (правый рисунок снизу), что является критичной проблемой для задачи сборки. Попробуем разобраться почему.
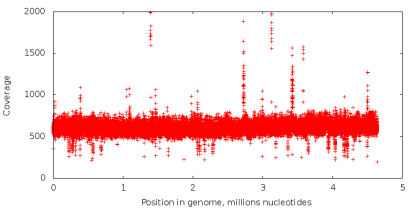
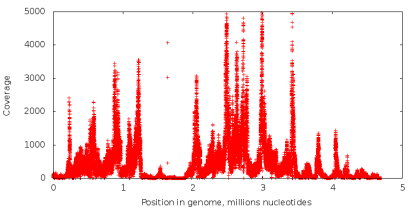
Проведем следующий эксперимент. Возьмем данные, полученные с помощью стандартного секвенирования, и из всех чтений извлечем подстроки фиксированной длины k (дальше — k-меры), так что с каждой возможной позиции в чтении начинается один k-мер (таким способом последовательность ACGTAC разбивается на четыре 3-мера: ACG, CGT, GTA и TAC). Далее, посчитаем, сколько раз каждый k-мер встречается в наших чтениях. Если некоторый k-мер появляется достаточно большое число раз (то есть имеет высокое покрытие), есть основание предположить, что он встречается и в исходном геноме, который мы секвенируем. Если же k-мер встречается редко, то он, скорее всего, содержит неверно считанный нуклеотид. Ошибки секвенирования являются случайным событием, и вероятность того, что одинаковая ошибка встретится сразу в большом количестве k-меров очень мала (вероятность неверного прочтения отдельно взятого нуклеотида колеблется от 0.001% до 1% в зависимости от позиции нуклеотида в чтении). Таким образом, с помощью величины покрытия k-меров, можно разделить их на достоверные и ошибочные. Подобные методы используются в большинстве современных ассемблеров и утилит для исправления ошибок в чтениях. Однако, в случае секвенирования единичной клетки, разделить k-меры подобным образом невозможно, так как k-меры, встречающиеся редко, могут соответствовать участку, скопированному небольшое число раз, и вовсе не содержать ошибок.
Неравномерное покрытие является не единственной проблемой подхода MDA. При копировании ДНК от основной нити ответвляется новая одноцепочечная нить. Она случайным образом может склеиться с другой отпочковавшейся нитью, из-за чего рядом окажутся последовательности ДНК, которые соответствуют совершенно разным участкам в исходном геноме. На место такой ошибочной склейки снова могут сесть “копировальные машины” и размножить неверный участок. Тогда, в результате секвенирования мы получим чтения, содержащие последовательности из разных частей генома (химерические чтения). При обработке таких чтений геномный сборщик может неверно объединить две последовательности в одну. Если один неверно поставленный нуклеотид не является грубой ошибкой, то два неверно склеенных участка — намного более серьезная проблема, которая затрудняет дальнейший анализ.
Напоследок
Именно данные, полученные с помощью технологии MDA, стали основным предметом исследований в Лаборатории алгоритмической биологии в Санкт-Петербурге. Было разработано множество алгоритмов, которые позволили обойти проблемы неравномерного покрытия и химерических чтений. Эти алгоритмы были реализованы в геномном ассемблере SPAdes, который сегодня является лидером в области сборки бактериальных геномов и используется ведущими мировыми лабораториями (например, в Объединенном геномном институте в США — крупнейшем в мире центре по изучению бактериальных геномов).
Дальше я бы с радостью познакомил читателей с базовыми принципами современных стандартных ассемблеров, рассказал бы о новых подходах и алгоритмах, которые используются при сборке данных, полученных по единичной клетке, но, к сожалению, в одну статью это не помещается. Для тех, кому интересно узнать больше об алгоритмах сборки — внизу есть ссылки на популярные тексты и научные публикации по этой теме. Искренне надеюсь, что следующая подобная статья появится быстрее, чем в этот раз.
Для заинтересованных
- Функцию “копировальной машины” выполняет ДНК-полимераза Фи-29 — белок, способный реплицировать участки ДНК в случайных местах.
- Чтобы определить и устранить ошибки секвенирования в данных, полученных при помощи MDA, используются подходы основанные на, к примеру, кластеризации k-меров по расстоянию Хэмминга, топологических особенностях графа де Брюйна в местах ошибок.
- Поиск и удаление химерических соединений также в основном производится по топологии графа де Брюйна.
Список литературы
- P. E. C. Compeau and P. A. Pevzner. Genome Reconstruction: A Puzzle with a Billion Pieces (обзорная статья о сборке геномов)
- P.A. Pevzner, H. Tang, and M. Waterman. An Eulerian path approach to DNA fragment assembly. Proceedings of the National Academy of Science of the United States of America, 98:9748-9753, 2001 (первая статья о применении графа де Брюйна в задаче сборки генома)
- D. Zerbino, and E. Birney. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Research. 18 (2008), 821–829 (классический ассемблер Velvet)
- A. Bankevich, S. Nurk, et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. Journal of Computational Biology 19(5) (2012), 455-477 (SPAdes — ассемблер для сборки данных, полученных при помощи MDA)
