Автор: нейрофизиолог научно-просветительского проекта Фанерозой, Анастасия Маркова.
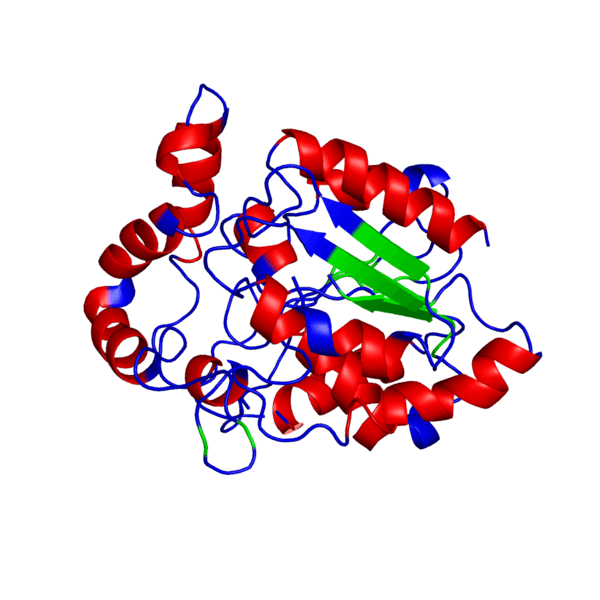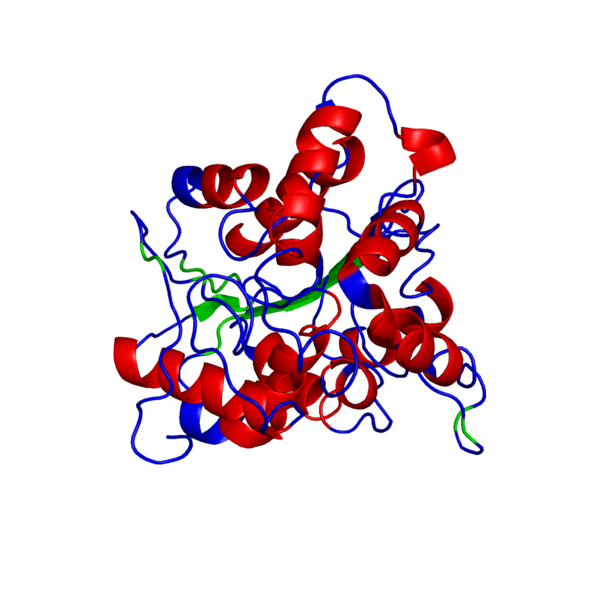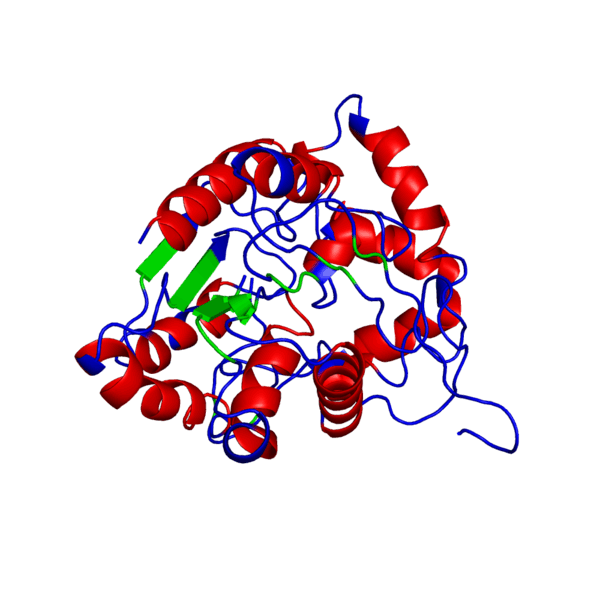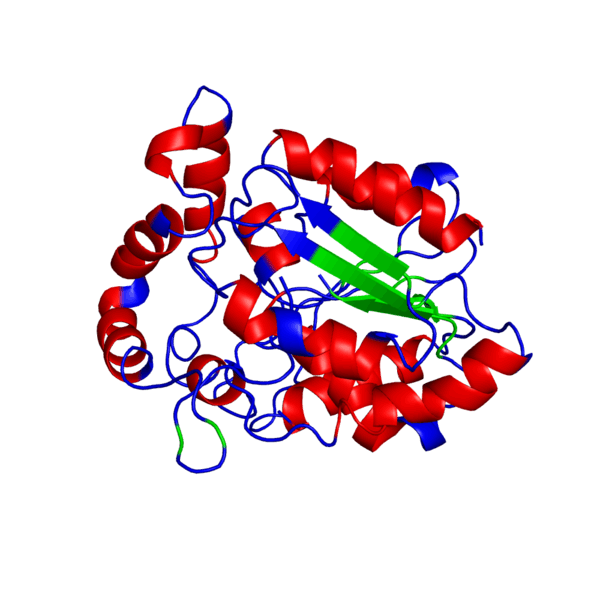
Наш мир полон разнообразия: повсюду встречаются различные виды растений и животных — повсюду кишит жизнь. И если так подумать, вспомнить школьные годы, то один из ученых, а именно Фридрих Энгельс когда-то дал свое особенное определение жизни, которое является одним из множества других:

В этом определении упоминаются некоторые “белковые тела”, а именно разнообразие химических соединений под названием “белок”. Белки, протеины или полипептиды — это высокомолекулярные органические вещества, состоящие из альфа-аминокислот, соединённых в цепочку пептидной связью. Белок — это сложное соединение, которое имеет несколько разных структур, которые определяются тем, какие именно связи задействованы в тот или иной момент для создания наиболее энергетически выгодной конформации данного соединения относительно второго закона термодинамики. У полипептидов существует четыре варианта укладки, четыре конформации или четыре структуры, в которых задействованы водородные, ионные, ковалентные, гидрофильно-гидрофобные связи, и таким образом, они образуют первичную, вторичную, третичную и четвертичную структуру.

В данной статье будет рассматриваться моделирование только одноцепочечных белков, так как формирование сложных белковых конгломератов — это уже отдельная тема.
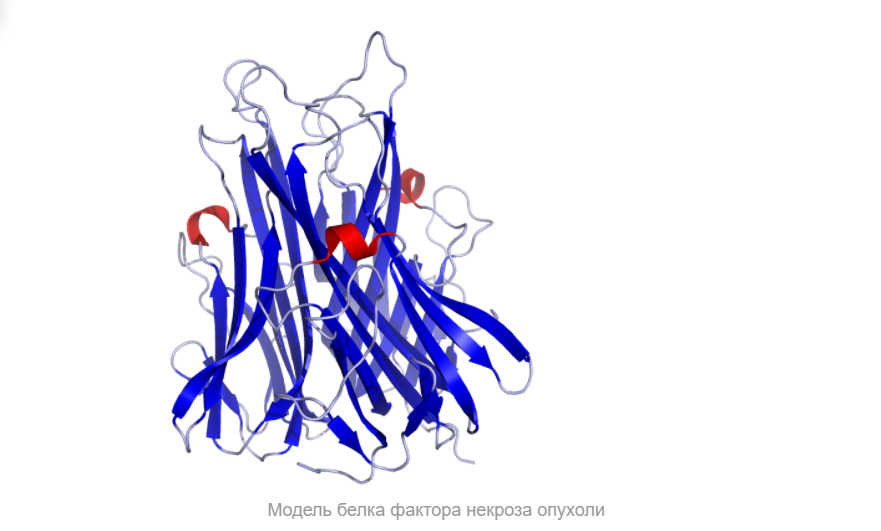
Он влияет на липидный метаболизм, коагуляцию, устойчивость к инсулину, функционирование эндотелия, стимулирует продукцию ИЛ-1, ИЛ-6, ИЛ-8, интерферона-гамма, активирует лейкоциты и является одним из важных факторов защиты от внутриклеточных паразитов и вирусов. Два рецептора TNF, которые принадлежат к семейству рецепторов фактора роста нервов с низким сродством, богатого цистеином (TNF-R1 и TNF-R2), являются единственными медиаторами передачи сигналов TNF. Считается, что передача сигналов происходит, когда тример TNF связывается с внеклеточными доменами двух или трех схожих рецепторных молекул, что делает возможной агрегацию и активацию цитоплазматических доменов.
Так уж эволюционно случилось, что белки у разных живых существ либо отличаются незначительно, либо отличаются довольно сильно. Тем не менее строение более консервативных участков и определенных последовательностей аминокислот, как правило, остается постоянной. Теперь, когда мы познакомились с объектом «нашего вожделения» нужно потихоньку приступать к делу.
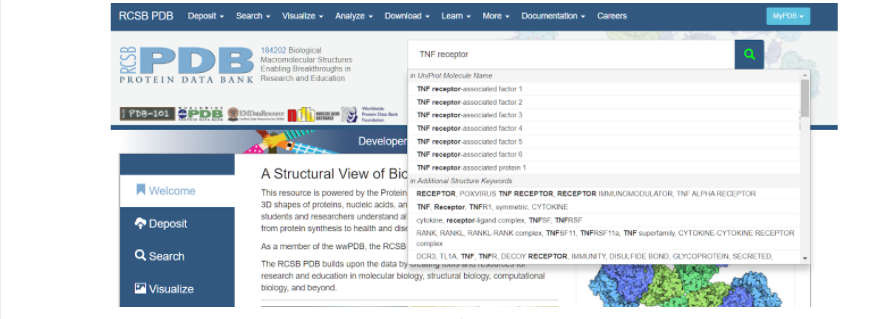
Итак, для того, чтобы начать моделирование, необходимо сначала найти исходный белок-матрицу в белковой базе данных (Protein Data Bank), на основании которого мы будем строить интересующий нас белок. В данном случае белком-матрицей будет выступать изученный и построенный TNFR человека. Для этого заходим на сайт RSCB Protein Data Bank и в поисковой строке вбиваем запрос: TNF receptor — и нажимаем на значок лупы несколько правее вкладки поиска модели белка.
После того как поиск закончен, можно наблюдать список самых разных моделей, построенные по различным методам. При выборе модельного белка стоит обратить внимание на качество сборки его модели — метод получения модели (электронная микроскопия или рентгеноструктурный анализ) и ее разрешение (количество ангстрем). При изучении списка найденных смоделированных протеинов стоит обратить внимание именно на те, которые получены методом рентгеноструктурного анализа, так как именно он позволяет создать модель молекулы более полную, а не только ее внешнее строение.
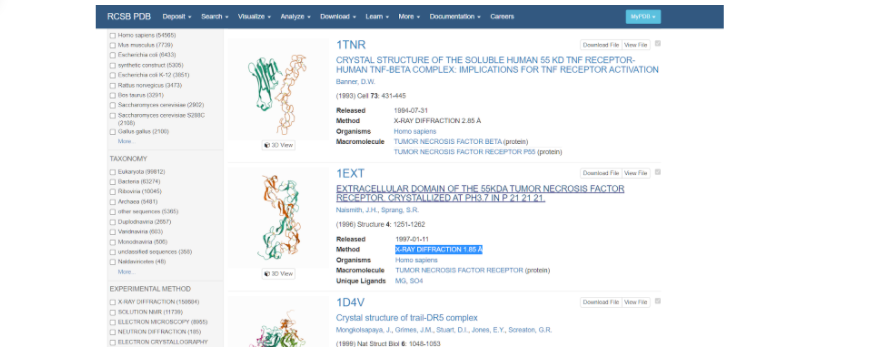
Выбираем ту модель белка, у которой наиболее высокое разрешение — здесь это модель под названием 1EXT. Нажимаем на название модели и переходим на ее основную страницу.
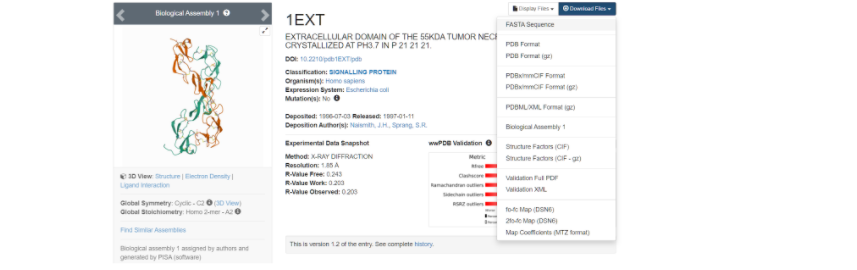
Здесь необходимо скачать файл FASTA Sequence в формате .txt для дальнейшей работы с фаста-файлом.

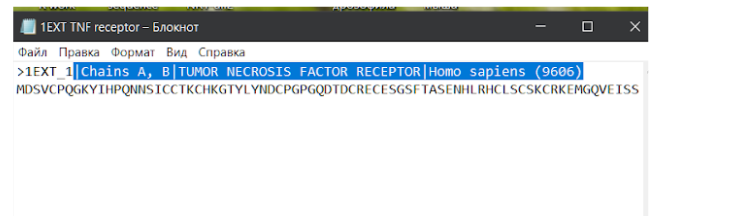
В сохраненном текстовом файле с аминокислотной последовательностью модельного исходного белка можно убрать лишние данные из названия, которые находятся в первой строке после значка “>”. Слишком длинные данные будут мешать при дальнейшей работе с другими средствами для поиска данных, выравнивания последовательностей или моделирования.
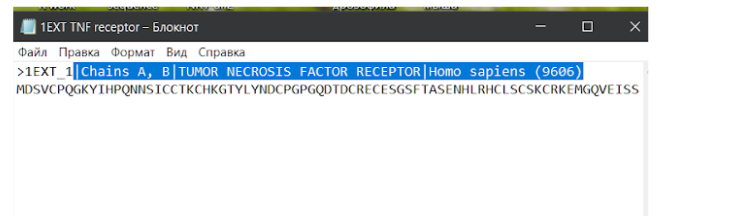
Теперь, после того, как мы определились с исходным белком, на основе которого будем строить модель нашего белка интереса, мы идем на сайт NCBI и там переходим на страницу алгоритма BLAST.
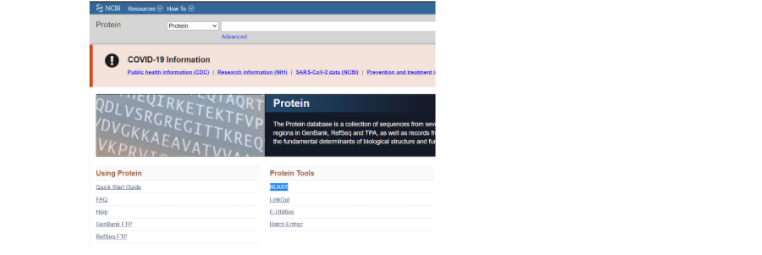
На данной странице стоит выбрать окошко, где большими буквами написано “Protein BLAST” для того, чтобы найти гомологичную последовательность для нашего модельного белка. Из этой гомологичной последовательности аминокислот мы и будем строить наш белок.
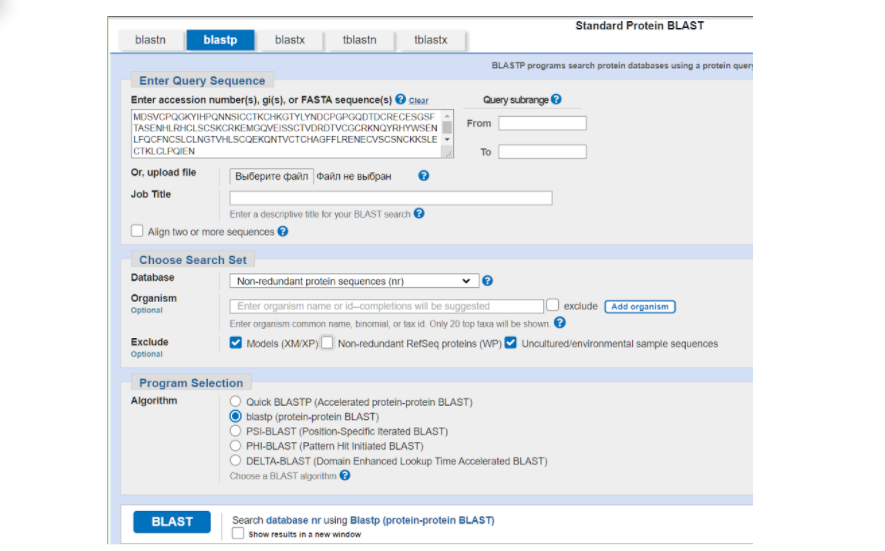
На странице BLASTа вставляем в большое окошко сиквенс нашего белка в FASTA формате из того текстового файла, который мы сохранили. Дальше выбираем те параметры, которые нас больше устраивают для поиска гомологичных последовательностей в БД. В графе DataBase можно выбрать один из интересующих нас вариантов массивов данных, в которых будет осуществляться поиск:
Для поиска гомологов чаще всего используется первый вариант. Дальше в графе “организм” вы можете ввести латинское название того организма, в котором возможно был найден гомологичный белок. Можно ввести название конкретного вида, а можно определенный таксон — насекомые, млекопитающие и др. или же, наоборот, исключить из рассмотрения тот или иной вид, или таксон. Например, я выбрала бархатистую летучую мышь Molossus molossus. Далее ставим галочки на исключение модельных последовательностей и некультивируемых проб и нажимаем на заветную кнопочку “BLAST”.
Страница несколько раз обновится, пока алгоритм будет выискивать наиболее подходящую последовательность.
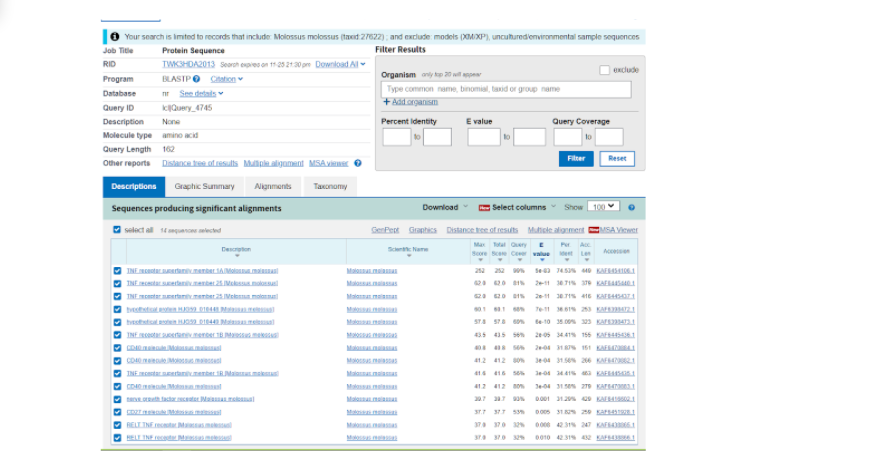
Далее нас перебрасывает на страницу, где уже представлены различные данные по тем последовательностям, которые более всего подходят к тому белку, который мы выбрали модельным. Здесь нас интересует та последовательность, которая в итоге показала наибольшее соответствие исходному белку и показала наибольший итоговый результат по сравнению с другими вариантами. Как правило, такие последовательности находятся в начале списка, а в данном случае вообще на первом месте.
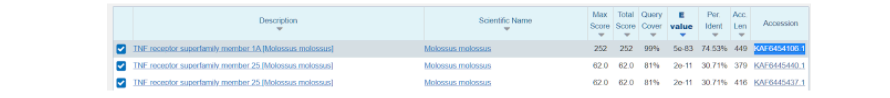
Здесь же во вкладке “Alignments”, или Выравнивания, мы можем посмотреть соответствие модельной последовательности и искомой. Из приведенных данных видно, что идентичность последовательностей составляет 75%, и исходя из этого можно воспользоваться “гомологическим моделированием” протеина.

Для того чтобы приступить к дальнейшим действиям, нам необходимо кликнуть на ID номер последовательности и перейти на страницу, которая уже содержит информацию об аминокислотной последовательности одной цепи внеклеточного рецептора фактора некроза опухоли.

На этой странице необходимо нажать на плашечку FASTA и перейти на страницу с последовательностью в FASTA формате, чтобы скачать оттуда, или можно поступить несколько проще и сразу на этой странице нажать на кнопку “отправить”, выбрать формат файла “FASTA”. Опять же нам необходимо скачать этот файл в формате .txt, чтобы продолжить работу. Это связано с тем, что формат FASTA на ПК в блокноте не открывается и не подлежит какому-либо дальнейшему редактированию. Даже в программе MEGA X, которая призвана работать с таким файлом, не получится адекватно редактировать его, даже при помощи ножниц. Итак…
После того, как сиквенс скачен в нужном формате, нам необходимо объединить две последовательности в один блокнот.
Первой должна быть последовательность интересующего нас белка, а второй — белка-модели. При этом, чтобы в дальнейшем файл правильно читался, нужно сделать так, чтобы две разные последовательности разделялись ↪ переносом строки. Теперь сохраняем файл и идем на сайт для выравнивания по методу ClustalW. И здесь стоит сделать еще одно лирическое отступление…
Теперь на сайте Multiple Sequence Alignment by CLUSTALW выбираем файл с двумя сиквенсами и выбираем настройки парного выравнивания SLOW/ACCURATE и нажимаем “выравнивать”.
После выравнивания перед вами предстанет страница с самим выравниванием, где будут показаны гомологичные аминокислоты и места в них, где сходства обнаружено не было. Эти места ещё называют на сленге “гэпы”, они же показаны значками “─”.

А теперь необходимо в новый файл блокнота скопировать только те места интерисующей нас последовательности, где нет “гэпов”, без пробелов и переносов строк. Так мы укоротим последовательность и облегчим последующую работу программы, которая поможет нам в составлении модели белка. Может возникнуть правильный вопрос: почему мы “отрезаем” такие большие части белка, ведь они могут совсем иначе сложиться, и в итоге белок будет не похож на тот, что мы используем в качестве модели? Дело все в том, что в организме в процессе фолдинга белка и его созревания принимают участие различные механизмы (происходит так называемая посттрансляционная модификация), которые, в том числе обрезают лишние части этого самого белка для того, чтобы в итоге он принял свою окончательную форму и работал правильно (ограниченный протеолиз).

В новом файле должны снова находиться обе последовательности в том же порядке: первой АК последовательности нас интересующей, а второй — модельной. Этот файл можно оставить в формате .txt, а можно при сохранении перевести в формат .fasta. Тут уж как вам захочется, так как здесь мы вышли уже на финишную прямую. Для моделирования белка нужно будет перейти на сайт со свободной возможностью смоделировать ваш белок методом гомологического моделирования — SWISS-MODEL.

Здесь мы нажимаем “начать моделирование” и нас перебрасывает на страницу, где необходимо выбрать различные настройки того, каким же образом мы будем осуществлять нашу затею.
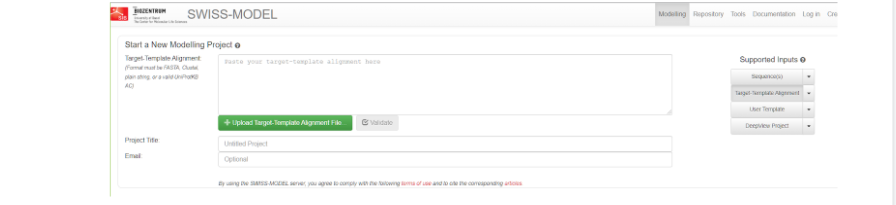
Здесь нужно выбрать справа параметр Target-Template Alignment, которое означает, что выравнивание матрицы и мишени задается пользователем. А в большое поле вы можете либо скопировать обе ваши последовательности из блокнота, либо вставить фаста-файл. После того как файл будет вставлен, вам выдаст пару или несколько вариантов возможного моделирования белка с теми последовательностями, которые оказались похожими в базе данных на нашу модель, но для более правильного моделирования стоит все же выбрать тот модельный белок, который мы выбрали заранее.
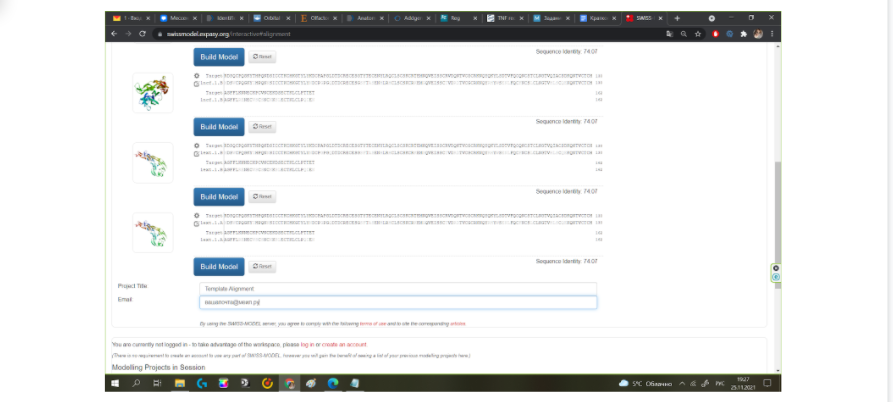
В выданном нам списке необходимый нам вариант моделирования находится предпоследним и последним, так как у внеклеточного рецептора фактора некроза опухоли две цепи — А и В, которые являются одинаковыми, но при укладке цепь “В” находится в антипараллельном положении к цепи “А”. В данном случае не имеет значения, какой из двух вариантов модельного белка 1EXT.1 вы выберите. Далее необходимо вписать название проекта и для удобства вписать свою почту, чтобы по окончании моделирования ваш проект был отправлен вам на почту, так как иногда при большой загруженности сервера процесс моделирования может занять несколько часов. Когда все данные введены, можно нажать на кнопку “построить модель” и подождать некоторое время.
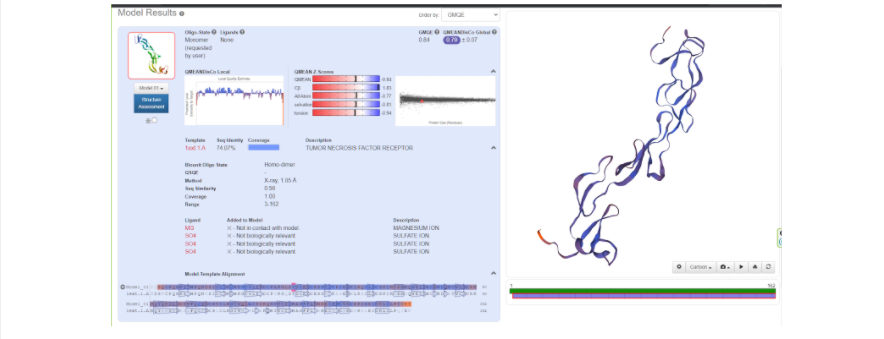
Когда же будет готово перед вами появится тот самый белочек, который вы хотели увидеть и потрогать, “покрутить в своих ручках”. В окне справа модель протеина как раз можно покрутить, посмотреть на него с разных сторон, а также поиграться с настройками, что именно показать на самой модели: серин-треониновые участки, альфа-спирали и бета-листы, гидрофобные участки, полярные области, и многое другое. Также слева приведены данные, которые соответствуют вашей модели:
QMEAN включает в себя 4 параметра:
Но это ещё не все данные, поэтому давайте рассмотрим оставшиеся:
А также ниже приведено выравнивание белка-мишени и белка-матрицы, построенное автоматически и отображенное графически. Каждому участку выравнивания приписывается определенное значение QMEAN (наилучшее качество — синий цвет, наихудшее — красный). Также для каждого участка матрицы и мишени указана вторичная структура (стрелки — это бета-листы, прямоугольники — альфа-спирали).
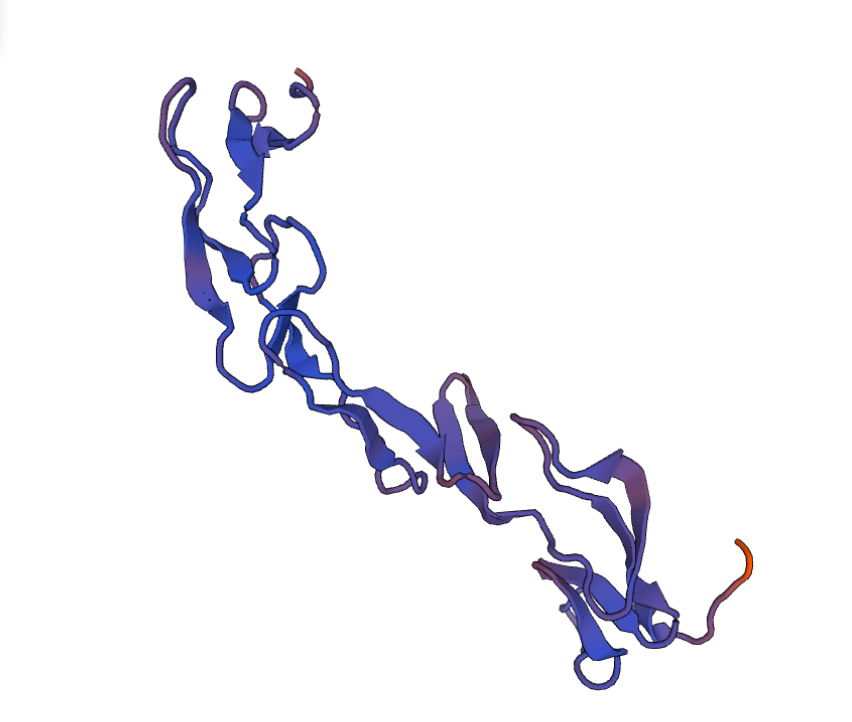
И вот на этой прекрасной ноте, после того, как моделирование белка было завершено, мне хотелось бы закончить данный фолиант и пожелать вам удачи в освоении новых горизонтов, даже таких сложных, как моделирование белковых молекул со всеми подводными камнями. Всем спасибо!

Благодарность: искренне благодарим биоинформатика Жукову Алину Александровну (к.б.н., доцент кафедры анатомии и физиологии человека и животных РГПУ им. А.И. Герцена) за дельные советы, проверку статьи и помощь
Дисклеймер от мистера ДНК- очевидность
Данная статья будет интересна тем, кто хоть как-то знаком с молекулярной биологией и биохимией, а также интересуется возможностью визуализации белков.
Данная статья будет интересна тем, кто хоть как-то знаком с молекулярной биологией и биохимией, а также интересуется возможностью визуализации белков.
Наш мир полон разнообразия: повсюду встречаются различные виды растений и животных — повсюду кишит жизнь. И если так подумать, вспомнить школьные годы, то один из ученых, а именно Фридрих Энгельс когда-то дал свое особенное определение жизни, которое является одним из множества других:
“Жизнь есть способ существования белковых тел, существенным моментом которого является постоянный обмен веществ с окружающей их внешней природой, причем с прекращением этого обмена веществ прекращается и жизнь, что приводит к разложению белка”.

В этом определении упоминаются некоторые “белковые тела”, а именно разнообразие химических соединений под названием “белок”. Белки, протеины или полипептиды — это высокомолекулярные органические вещества, состоящие из альфа-аминокислот, соединённых в цепочку пептидной связью. Белок — это сложное соединение, которое имеет несколько разных структур, которые определяются тем, какие именно связи задействованы в тот или иной момент для создания наиболее энергетически выгодной конформации данного соединения относительно второго закона термодинамики. У полипептидов существует четыре варианта укладки, четыре конформации или четыре структуры, в которых задействованы водородные, ионные, ковалентные, гидрофильно-гидрофобные связи, и таким образом, они образуют первичную, вторичную, третичную и четвертичную структуру.
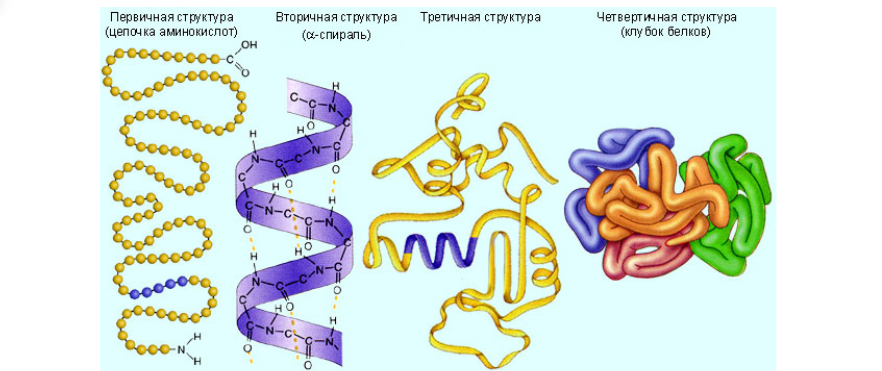
В данной статье будет рассматриваться моделирование только одноцепочечных белков, так как формирование сложных белковых конгломератов — это уже отдельная тема.
Итак, что же нужно для того, чтобы немного “покрутить в своих ручках” белок, которого наверняка нет в базе данных и который гипотетически можно было бы использовать в различных целях в будущем?Очевидно, что для начала его нужно самостоятельно построить, а потому первым делом нам следует определиться, какой же белок мы хотим смоделировать. В данном случае мой выбор пал на весьма интересный белок — внеклеточный рецептор фактора некроза опухоли (TNFR, ФНОР). Сам же белок фактора некроза опухоли (ФНО), как вы уже поняли из его названия, является внеклеточным белком, являющимся многофункциональным провоспалительным цитокином, синтезирующимся в основном моноцитами и макрофагами.
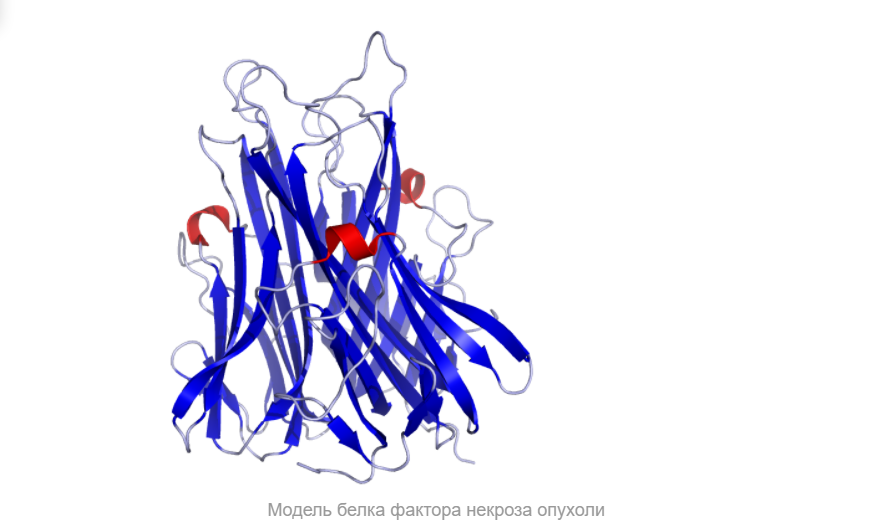
Он влияет на липидный метаболизм, коагуляцию, устойчивость к инсулину, функционирование эндотелия, стимулирует продукцию ИЛ-1, ИЛ-6, ИЛ-8, интерферона-гамма, активирует лейкоциты и является одним из важных факторов защиты от внутриклеточных паразитов и вирусов. Два рецептора TNF, которые принадлежат к семейству рецепторов фактора роста нервов с низким сродством, богатого цистеином (TNF-R1 и TNF-R2), являются единственными медиаторами передачи сигналов TNF. Считается, что передача сигналов происходит, когда тример TNF связывается с внеклеточными доменами двух или трех схожих рецепторных молекул, что делает возможной агрегацию и активацию цитоплазматических доменов.
Так уж эволюционно случилось, что белки у разных живых существ либо отличаются незначительно, либо отличаются довольно сильно. Тем не менее строение более консервативных участков и определенных последовательностей аминокислот, как правило, остается постоянной. Теперь, когда мы познакомились с объектом «нашего вожделения» нужно потихоньку приступать к делу.
Итак, для того, чтобы начать моделирование, необходимо сначала найти исходный белок-матрицу в белковой базе данных (Protein Data Bank), на основании которого мы будем строить интересующий нас белок. В данном случае белком-матрицей будет выступать изученный и построенный TNFR человека. Для этого заходим на сайт RSCB Protein Data Bank и в поисковой строке вбиваем запрос: TNF receptor — и нажимаем на значок лупы несколько правее вкладки поиска модели белка.
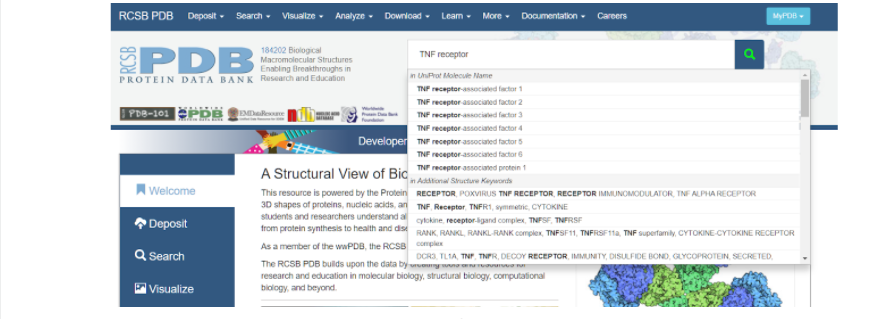
После того как поиск закончен, можно наблюдать список самых разных моделей, построенные по различным методам. При выборе модельного белка стоит обратить внимание на качество сборки его модели — метод получения модели (электронная микроскопия или рентгеноструктурный анализ) и ее разрешение (количество ангстрем). При изучении списка найденных смоделированных протеинов стоит обратить внимание именно на те, которые получены методом рентгеноструктурного анализа, так как именно он позволяет создать модель молекулы более полную, а не только ее внешнее строение.
Сейчас может возникнуть резонный вопрос, почему рекомендуется построение белка на основе уже известной модели, а не просто известной аминокислотной последовательности?Но это было лирическое отступление, а теперь — продолжим:
Дело в том, что большая часть алгоритмов предсказывающих различные структуры белка, опираются только на физические принципы. Таким образом алгоритмы пытаются воспроизвести межатомные взаимодействия в белковой молекуле и определить совместимую энергию, присущую любой возможной конформации данного белка.
В вычислительном аспекте проблема предсказания структуры белка выглядит как задача поиска глобального минимума функций совместимой энергии конформаций. Пока что этот подход не сильно преуспел: частично в силу несостоятельности выведенной функции энергии и частично ввиду того, что известные на сегодняшний день алгоритмы минимизации неизбежно застревают в локальных минимумах.
Альтернативу априорным методам представляет подход, предполагающий восстановление целостной картины структуры белка, путем поиска последовательностей, образующих подобные ему структуры. Методы, которые объединяет в себе этот подход, эмпирические, то есть основаны на опыте. Создаются алгоритмы, построенные на механизмах анализа знаний полученных во время биохимических опытов. Эти алгоритмы также пытаются предсказывать структуру белка на основании информации, почерпнутой из базы данных известных структур. Есть различные методы предсказания структуры интересующего белка: сравнительное моделирование (чем мы с вами и займемся), распознавание сверток, предсказания вторичной структуры, предсказания ab initio и др.
Если последовательность белка неизвестной структуры выровнена с одной или несколькими последовательностями белков с известной структурой (что можно сделать, например, в программе MEGA X) и в выравнивании 80 или более остатков показывает в лучшем случае 25% подобия, то средства множественного выравнивания позволяют предсказать структуру, принимаемую целевой последовательностью, на основании сравнения с известной (эталонной) структурой. Такой метод называют сравнительным моделированием или моделированием гомологии. Он дает возможность построить полную модель расположения атомов третичной структуры.
Если подходящие эталонные структуры для данной целевой последовательности не существуют, то остается прибегнуть к альтернативному подходу — предсказанию вторичной структуры. Этот путь ведет к предсказанию свойственного каждому остатку состояния вторичной структуры: спирального, нитевидного, листовидного или катушкообразного. Такие предсказания иногда называют предсказаниями трех состояний.
Методы распознавания сверток (альфа-считывания) позволяют обнаружить отдаленные отношения и отделить их от случайных подобий последовательностей, не связанных с общей свертой. Разработанные на их основе алгоритмы осуществляют поиск в библиотеке известных структур белка и находят структуру, наиболее подходящую для запрашиваемой последовательности, структура которой и должна быть предсказана. После построения выравнивания между последовательностью запроса и отдаленно связанными последовательностями из базы данных может быть получена полная картина искомой трехмерной структуры белка.
Методы ab initio предполагают предсказание структуры белков от первых принципов и опираются на различные теории физических наук, например статистической термодинамики и квантовой механики. И из всех этих методов самым точным и всесторонним является сравнительное моделирование.
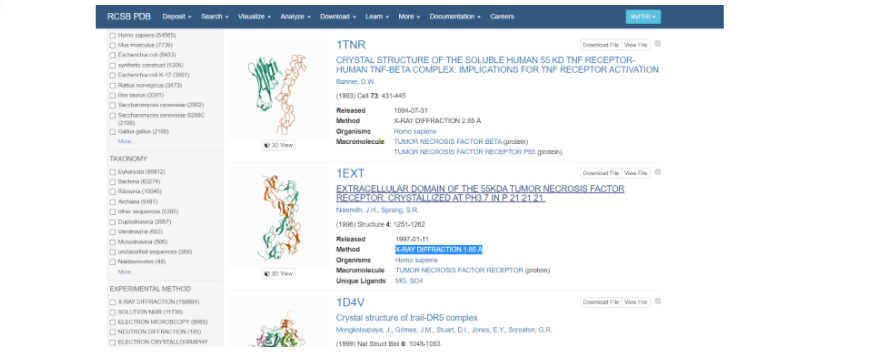
Выбираем ту модель белка, у которой наиболее высокое разрешение — здесь это модель под названием 1EXT. Нажимаем на название модели и переходим на ее основную страницу.
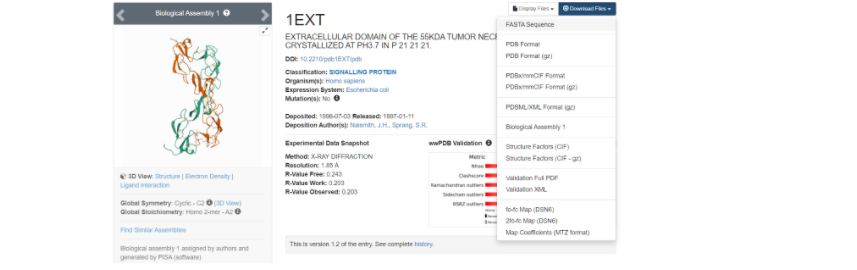
Здесь необходимо скачать файл FASTA Sequence в формате .txt для дальнейшей работы с фаста-файлом.
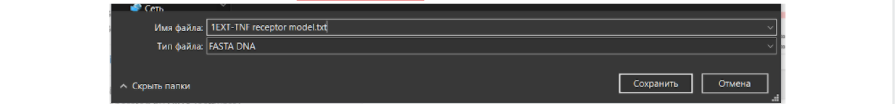
В сохраненном текстовом файле с аминокислотной последовательностью модельного исходного белка можно убрать лишние данные из названия, которые находятся в первой строке после значка “>”. Слишком длинные данные будут мешать при дальнейшей работе с другими средствами для поиска данных, выравнивания последовательностей или моделирования.
Теперь, после того, как мы определились с исходным белком, на основе которого будем строить модель нашего белка интереса, мы идем на сайт NCBI и там переходим на страницу алгоритма BLAST.
На данной странице стоит выбрать окошко, где большими буквами написано “Protein BLAST” для того, чтобы найти гомологичную последовательность для нашего модельного белка. Из этой гомологичной последовательности аминокислот мы и будем строить наш белок.
Что такое вообще BLAST?В нашем случае мы будем пользоваться пакетом BLASTP, для поиска гомологичных последовательностей к исходному белку.
Basic Local Alignment Search Tool — основное (программное) средство, или семейство алгоритмов поиска локальных выравниваний. Заблудившиеся в научной мысли люди, иногда обитающие в комментариях, снова могут сказать, что на картинке NCBI BLAST и он к BLAST никакого отношения не имеет. Дескать это всего лишь веб-интерфейс. Ребята, называйте NCBI BLAST хоть «маной небесной», но попадая на сайт и используя эту «ману небесную» в своих целях, вы автоматически работаете с алгоритмом BLAST. Поэтому далее мы будем называть бласт на NCBI — бластом вопреки мнениям некоторых людей. Не нравится — пишите опровержение, а не засоряйте комментарии своей токсичностью, ведь это бесмыссленно.
Итак, программное средство Basic Local Alignment Search Tool было написано Альтшулем с сотрудниками в 1990 году. Благодаря своей эффективности и развитому статистическому аппарату, оно снискало себе широкую популярность. В пакет BLAST входят программы для нахождения локального выравнивания с высоким весом между данной последовательностью и последовательностями из базы данных, как для ДНК, так и для белковых последовательностей.
Идея, лежащая в основе алгоритма BLAST, состоит в том, что правильное выравнивание наверняка будет содержать в себе короткий участок подряд идущих одинаковых остатков, или участок с очень высоким весом. Следовательно, сначала мы можем искать в базе данных только короткие совпадения, а затем использовать их как “затравки”, из которых путем расширения начальных совпадений получать более длинное хорошее выравнивание. Условие коротких затравок дает возможность заранее обработать данную последовательность, чтобы сделать таблицу всех возможных затравок с их координатами в нашей последовательности.
BLAST создает список всех “близких” слов фиксированной длины (по умолчанию 3 для белковых последовательностей, 11 — для нуклеотидных), которые бы локально выравнивались с нашей последовательностью с весом, выше некого порогового значения, обычно около 2 бит на остаток. Затем алгоритм сканирует базу данных, и каждый раз при нахождении слова из списка начинает процесс “расширения совпадения”, чтобы увеличить возможный участок выравнивания без разрывов или так называемых “гэпов”, в обоих направлениях, до достижения максимального веса.
Наиболее широко используется только безразрывное выравнивание, так как при таком ограничении алгоритм теряет только малую часть качественных выравниваний, потому что ожидаемый наилучший вес неродственных последовательностей быстро падает, в то время как веса безразрывных выравниваний фрагментов родственных последовательностей все еще могут быть значительными.
В алгоритме поиска BLAST имеется несколько пакетов: BLASTP (сравнивает аминокислотную последовательность запроса с предметными последовательностями данных белка), BLASTN (сравнивает запрашиваемую нуклеотидную последовательность с последовательностями из БД), BLASTX (сравн. результаты машинной смысловой трансляции с шестью рамками (обеих нитей) последовательности запроса нуклеотидов с БД белков), TBLASTN (сравн. белковую последовательность запроса с последовательностями из базы данных нуклеотидных последовательностей, динамически транслируемых с шестью рамками считывания (обе нити) и PSI-BLAST (сравн. АК последовательность запроса с предметными последовательностями из базы данных белка).
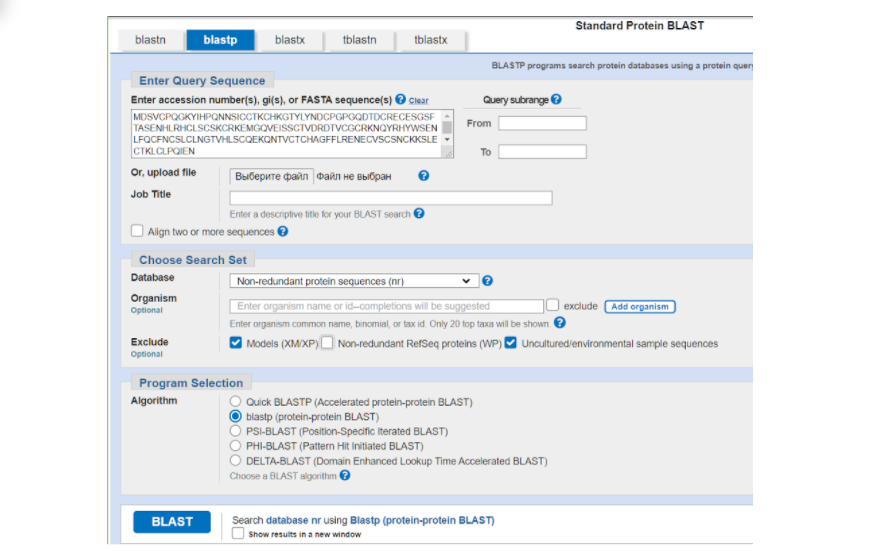
На странице BLASTа вставляем в большое окошко сиквенс нашего белка в FASTA формате из того текстового файла, который мы сохранили. Дальше выбираем те параметры, которые нас больше устраивают для поиска гомологичных последовательностей в БД. В графе DataBase можно выбрать один из интересующих нас вариантов массивов данных, в которых будет осуществляться поиск:
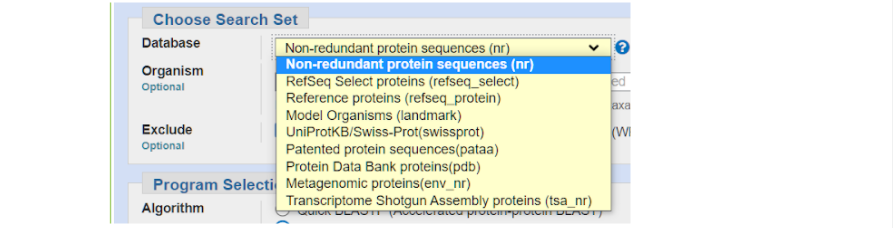
- Неповторяющиеся (non-redundant) белковые последовательности (из баз данных GenBank, PDB, Swiss Prot и др. — по умолчанию; наибольшее число вариантов).
- Только последовательности белков, аннотированные в NCBI (Refseq_protein).
- Model Organisms (протеомы 27 геномов, перекрывающих различные таксоны — число белков существенно меньше; используется вместе с методом SmartBLAST.)
- UniProtKB/Swiss-Prot (надежная информация о белковых последовательностях).
- Patented protein sequences (только запатентованные коммерческие последовательности белков).
- Protein Data Bank proteins (белки, представленные в базе данных PDB, для которых экспериментально установлена первичная и трехмерная структуры).
- Metagenomic proteins (последовательности получены методами метагеномики — совместное секвенирование геномов экологической группы организмов, обитающих в тех или иных условиях).
- Transcriptome Shotgun Assembly proteins (информация о белках получена на основе транскриптомов, исследованных фрагментарно).

Для поиска гомологов чаще всего используется первый вариант. Дальше в графе “организм” вы можете ввести латинское название того организма, в котором возможно был найден гомологичный белок. Можно ввести название конкретного вида, а можно определенный таксон — насекомые, млекопитающие и др. или же, наоборот, исключить из рассмотрения тот или иной вид, или таксон. Например, я выбрала бархатистую летучую мышь Molossus molossus. Далее ставим галочки на исключение модельных последовательностей и некультивируемых проб и нажимаем на заветную кнопочку “BLAST”.
Страница несколько раз обновится, пока алгоритм будет выискивать наиболее подходящую последовательность.
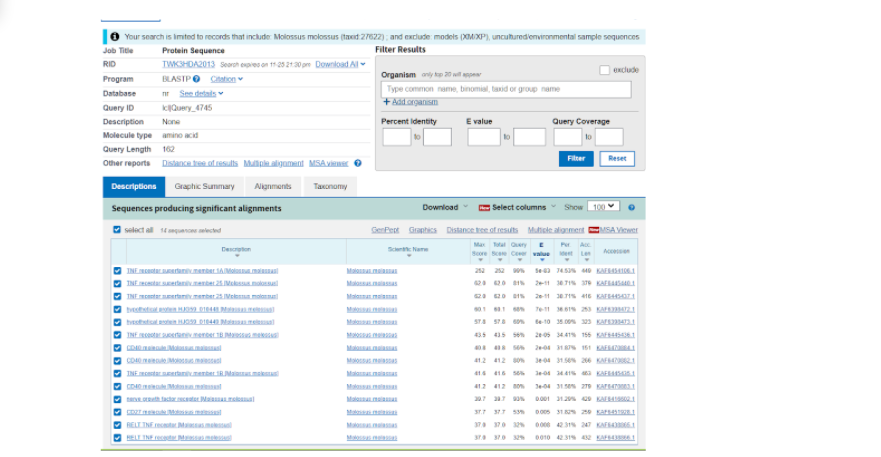
Далее нас перебрасывает на страницу, где уже представлены различные данные по тем последовательностям, которые более всего подходят к тому белку, который мы выбрали модельным. Здесь нас интересует та последовательность, которая в итоге показала наибольшее соответствие исходному белку и показала наибольший итоговый результат по сравнению с другими вариантами. Как правило, такие последовательности находятся в начале списка, а в данном случае вообще на первом месте.
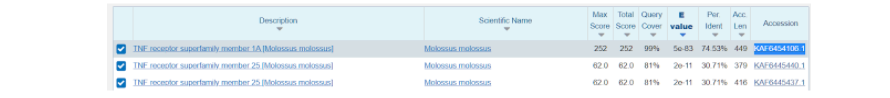
Здесь же во вкладке “Alignments”, или Выравнивания, мы можем посмотреть соответствие модельной последовательности и искомой. Из приведенных данных видно, что идентичность последовательностей составляет 75%, и исходя из этого можно воспользоваться “гомологическим моделированием” протеина.

Для того чтобы приступить к дальнейшим действиям, нам необходимо кликнуть на ID номер последовательности и перейти на страницу, которая уже содержит информацию об аминокислотной последовательности одной цепи внеклеточного рецептора фактора некроза опухоли.

На этой странице необходимо нажать на плашечку FASTA и перейти на страницу с последовательностью в FASTA формате, чтобы скачать оттуда, или можно поступить несколько проще и сразу на этой странице нажать на кнопку “отправить”, выбрать формат файла “FASTA”. Опять же нам необходимо скачать этот файл в формате .txt, чтобы продолжить работу. Это связано с тем, что формат FASTA на ПК в блокноте не открывается и не подлежит какому-либо дальнейшему редактированию. Даже в программе MEGA X, которая призвана работать с таким файлом, не получится адекватно редактировать его, даже при помощи ножниц. Итак…
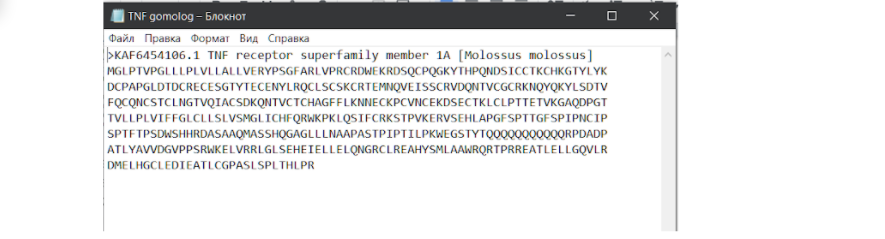
После того, как сиквенс скачен в нужном формате, нам необходимо объединить две последовательности в один блокнот.

Первой должна быть последовательность интересующего нас белка, а второй — белка-модели. При этом, чтобы в дальнейшем файл правильно читался, нужно сделать так, чтобы две разные последовательности разделялись ↪ переносом строки. Теперь сохраняем файл и идем на сайт для выравнивания по методу ClustalW. И здесь стоит сделать еще одно лирическое отступление…
? Алгоритм ClustalW является классическим алгоритмом прогрессивного множественного выравнивания и выполняется следующим образом:
- сначала рассчитывается матрица расстояния между последовательностями либо быстрым методом, считающим совпадения пар АК остатков или коротких нуклеотидных фрагментов (2-4 основания), либо классическим алгоритмом глобального выравнивания последовательностей с типичными штрафами за пропуски;
- после строится направляющее дерево guide tree методом присоединения соседей (neighbor joining), укореняя его методом “средней точки” — поиска позиции, в которой средние длины ветвей по обе стороны от корневого узла равны;
- и затем построение множественного выравнивания происходит через серию парных выравниваний типа последовательность-последовательность, последовательность-профиль и профиль-профиль в соответствии с направляющим деревом. В качестве значений матрицы замещения для выравниваний, использующих профиль, применяется среднее значение возможных сочетаний остатков выравниваемых позиций.

Теперь на сайте Multiple Sequence Alignment by CLUSTALW выбираем файл с двумя сиквенсами и выбираем настройки парного выравнивания SLOW/ACCURATE и нажимаем “выравнивать”.
После выравнивания перед вами предстанет страница с самим выравниванием, где будут показаны гомологичные аминокислоты и места в них, где сходства обнаружено не было. Эти места ещё называют на сленге “гэпы”, они же показаны значками “─”.

А теперь необходимо в новый файл блокнота скопировать только те места интерисующей нас последовательности, где нет “гэпов”, без пробелов и переносов строк. Так мы укоротим последовательность и облегчим последующую работу программы, которая поможет нам в составлении модели белка. Может возникнуть правильный вопрос: почему мы “отрезаем” такие большие части белка, ведь они могут совсем иначе сложиться, и в итоге белок будет не похож на тот, что мы используем в качестве модели? Дело все в том, что в организме в процессе фолдинга белка и его созревания принимают участие различные механизмы (происходит так называемая посттрансляционная модификация), которые, в том числе обрезают лишние части этого самого белка для того, чтобы в итоге он принял свою окончательную форму и работал правильно (ограниченный протеолиз).
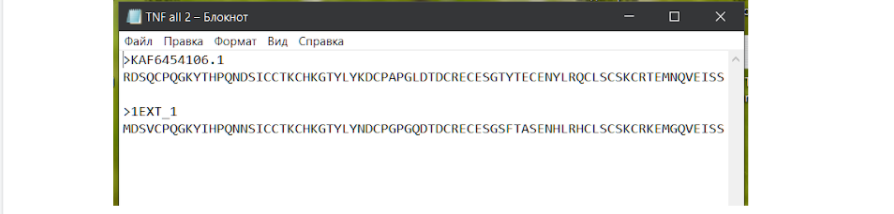
В новом файле должны снова находиться обе последовательности в том же порядке: первой АК последовательности нас интересующей, а второй — модельной. Этот файл можно оставить в формате .txt, а можно при сохранении перевести в формат .fasta. Тут уж как вам захочется, так как здесь мы вышли уже на финишную прямую. Для моделирования белка нужно будет перейти на сайт со свободной возможностью смоделировать ваш белок методом гомологического моделирования — SWISS-MODEL.

Здесь мы нажимаем “начать моделирование” и нас перебрасывает на страницу, где необходимо выбрать различные настройки того, каким же образом мы будем осуществлять нашу затею.
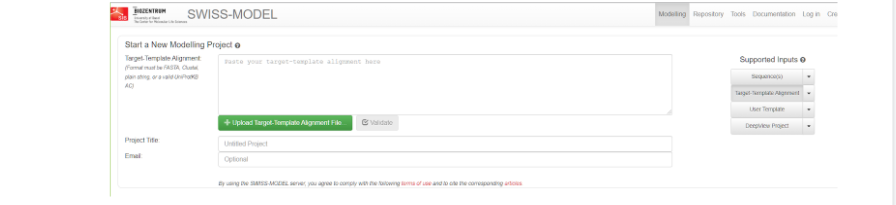
Здесь нужно выбрать справа параметр Target-Template Alignment, которое означает, что выравнивание матрицы и мишени задается пользователем. А в большое поле вы можете либо скопировать обе ваши последовательности из блокнота, либо вставить фаста-файл. После того как файл будет вставлен, вам выдаст пару или несколько вариантов возможного моделирования белка с теми последовательностями, которые оказались похожими в базе данных на нашу модель, но для более правильного моделирования стоит все же выбрать тот модельный белок, который мы выбрали заранее.

В выданном нам списке необходимый нам вариант моделирования находится предпоследним и последним, так как у внеклеточного рецептора фактора некроза опухоли две цепи — А и В, которые являются одинаковыми, но при укладке цепь “В” находится в антипараллельном положении к цепи “А”. В данном случае не имеет значения, какой из двух вариантов модельного белка 1EXT.1 вы выберите. Далее необходимо вписать название проекта и для удобства вписать свою почту, чтобы по окончании моделирования ваш проект был отправлен вам на почту, так как иногда при большой загруженности сервера процесс моделирования может занять несколько часов. Когда все данные введены, можно нажать на кнопку “построить модель” и подождать некоторое время.
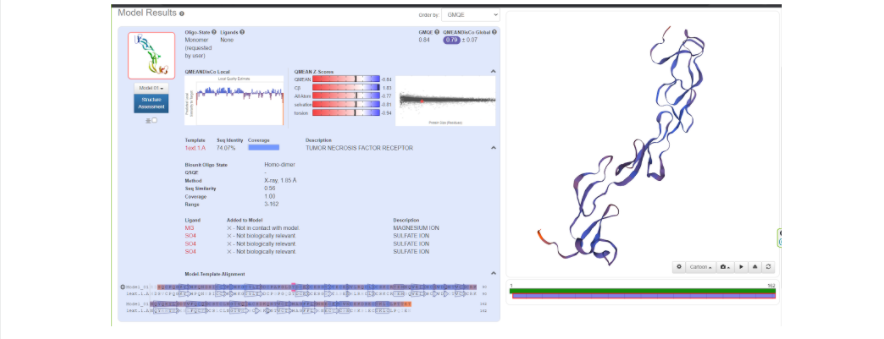
Когда же будет готово перед вами появится тот самый белочек, который вы хотели увидеть и потрогать, “покрутить в своих ручках”. В окне справа модель протеина как раз можно покрутить, посмотреть на него с разных сторон, а также поиграться с настройками, что именно показать на самой модели: серин-треониновые участки, альфа-спирали и бета-листы, гидрофобные участки, полярные области, и многое другое. Также слева приведены данные, которые соответствуют вашей модели:
- GMQE (Global Model Quality Estimation) — ожидаемое качество модели,
учитывающее выравнивание мишени с моделью и покрытии мишени
моделью. Варьирует от 0 до 1. - QMEAN — глобальная оценка нативности модели, основанная на
разнообразных геометрических характеристиках: сравнение их с таковыми в экспериментальных структурах белков подобного размера. Значение, близкое
к 0 — качество модели соответствует таковому для средней
экспериментальной структуры. Значения меньше -4.0 означают, что качество
модели низкое.
QMEAN включает в себя 4 параметра:
- Cbeta – оценка потенциалов взаимодействия между С-бета атомами,
- All atom – оценка потенциалов взаимодействия между всеми атомами,
- Solvation – оценка потенциалов сольватации,
- Torsion – оценка потенциалов торсионных углов.
Но это ещё не все данные, поэтому давайте рассмотрим оставшиеся:
- Local quality estimate – локальная оценка качества модели на уровне
отдельных аминокислотных остатков (<0.6 – низкое качество). - Сomparison – сравнение с нормированными значениями QMEAN для белков
с экспериментально известной структурой. Оценка дается в Z-value –
величине стандартных отклонений от среднего для белков определенного
размера. - Seq Identity — процент идентичности мишени и матрицы.
- Сoverage – область покрытия матрицей мишенью.
А также ниже приведено выравнивание белка-мишени и белка-матрицы, построенное автоматически и отображенное графически. Каждому участку выравнивания приписывается определенное значение QMEAN (наилучшее качество — синий цвет, наихудшее — красный). Также для каждого участка матрицы и мишени указана вторичная структура (стрелки — это бета-листы, прямоугольники — альфа-спирали).
И вот на этой прекрасной ноте, после того, как моделирование белка было завершено, мне хотелось бы закончить данный фолиант и пожелать вам удачи в освоении новых горизонтов, даже таких сложных, как моделирование белковых молекул со всеми подводными камнями. Всем спасибо!

Источники
1. National Center for Biotechnology Information — www.ncbi.nlm.nih.gov
2. pubmed.ncbi.nlm.nih.gov
3. Григорий Мавропуло-Столяренко, Александр Тулуб, Василий Стефанов. Биоинформатика. Учебник для академического бакалавриата.
4. Жуан Сетубал, Жуан Мейданис ВВЕДЕНИЕ В ВЫЧИСЛИТЕЛЬНУЮ МОЛЕКУЛЯРНУЮ БИОЛОГИЮ
5. Дурбин Р., Эдди Ш. Анализ биологических последовательностей
6. Игнасимуту С Основы биоинформатики
7. Модель TNF-R Molossus Molossus swissmodel.expasy.org/interactive/6KRqdR/models
2. pubmed.ncbi.nlm.nih.gov
3. Григорий Мавропуло-Столяренко, Александр Тулуб, Василий Стефанов. Биоинформатика. Учебник для академического бакалавриата.
4. Жуан Сетубал, Жуан Мейданис ВВЕДЕНИЕ В ВЫЧИСЛИТЕЛЬНУЮ МОЛЕКУЛЯРНУЮ БИОЛОГИЮ
5. Дурбин Р., Эдди Ш. Анализ биологических последовательностей
6. Игнасимуту С Основы биоинформатики
7. Модель TNF-R Molossus Molossus swissmodel.expasy.org/interactive/6KRqdR/models
